Senolytic drugs have been the most promising near-term anti-aging therapy since the ground-breaking paper by van Deursen of Mayo Clinic published in 2011. The body accumulates senescent cells as we age, damaged cells that send out signal molecules that in turn modify our biochemistry in a toxic, pro-inflammatory direction. Though the number of such cells is small, the damage they do is great. Van Deursen showed that just getting rid of these cells could increase lifespan of mice by ~25%. But he did it with a trick, using genetically engineered mice in which the senescent cells had a built-in self-destruct switch.
After that, the race was on to find chemical agents that would do the same thing without the genetically engineered self-destruct. They must selectively kill senescent cells, while leaving all other cells unharmed. It’s a tall order, because even a little residual toxicity to normal cells can be quite damaging. Before last week, the two best candidates were FOXO4-DRI and a combination of quercetin with dasatinib.
I’ve written in the past (here and here) that senolytic drugs are our best prospect for a near-term lift on the road to anti-aging medicine.
Last week, a large research group affiliated with the original May Clinic team published findings about fisetin, the latest and greatest candidate for a senolitic pill, another flavenoid, very close in structure to quercetin.
They grew senescent and normal cells in a test tube, then tested 11 different plant-derived chemicals for power to kill the one while leaving the other unharmed. The winner was fisetin.
(MEF stands for Mouse Embryonic Fybroblast, the cells that were cultured in the screening experiment.)
Fisetin is especially interesting because it is cheap, easily available, widely-regarded as safe, but not nearly as well studied as quercetin.
They took the winner, fisetin, and subjected it to a series of tests. They began with in vitro (cell culture) tests and proceeded to in vivo tests with live animals, culminating with an impressive life span assay in mice.
(The runner-up was curcumin, less interesting perhaps only because it has already been extensively studied. The curcumin molecule is unrelated to quercetin or fisetin, and is not a flavenoid. I can’t help but wonder if they had subjected curcumin to the same thorough testing that they reserved for fisetin, how would curcumin have fared?)
curcumin
The paper’s principal findings were:
Fisetin has lower liver toxicity (at equivalent doses for senolytic benefit) than any of the other senolytics tested so far.
Fisetin reduces pro-inflammatory signaling in a short course given to mice and in long-term experiments where fisetin was added to the mouse chow.
Fisetin reduces number of senescent fat cells in a short course given to mice.
Mice fed fisetin for long periods had much more glutathione than control mice. (Glutathione is one of very few marker molecules that seems to be wholly beneficial.)
Most impressively, mice fed fisetin late in life lived 10-15% longer than control mice. This represents a 50% increase in the remaining lifespan after the intervention.
What we know and what we’d really like to know
We’d like to know, do humans who take large doses of fisetin live longer? Do they have toxic side-effects? These questions require decades to answer.
Does fisetin reduce age markers in humans, especially methylation age? This is a feasible study, since the test is mature and safety of fisetin is fairly well established for short courses. Perhaps this experiment is being considered; I’ve written to the corresponding authors of the most recent study, in case they haven’t already thought of it. This test would not be definitive because we know that methylation age is not perfectly correlated with biological age; but if positive it would confirm both that fisetin is accomplishing epigenetic rejuvenation and that methylation tests were correctly informing us of this; a negative result would be ambiguous.
Episodic Dosing
It makes sense that senolytics should be taken periodically, not continuously. A high dose can be toxic to existing senescent cells, and then getting out of the way, it can allow normal cells to recover from any damage. This sounds like good theory, but different dosing regimens have not been tested experimentally. In fact, the new paper reports positive results from both high episodic dosing and lower everyday dosing.
The Mayo group had previously tested fisetin, and found it effective in killing some kinds of human senescent cells but not others. In previous tests, fisetin was found to be effective in senescent fat cells (pre-adipocyte, white adipose tissue), and that is where it was primarily tested in the new studies.
Authors’ comments
They note that the episodic treatment and short half-life suggest that the benefits of fisetin come from its senolytic action, rather than other actions as an antioxidant and signal molecule. They emphasize that clearing senescent white blood cells and making room for new, active white blood cells are activities that enhance the benefits of fisetin, since white blood cells contribute to clearing the remaining senescent cells.
Fisetin has previously been shown to have anti-cancer activity and to inhibit inflammatory signals directly. Here is a review of benefits of fisetin from three years ago. Drugage lists just two previous lifespan studies with fisetin, with encouraging results from yeast and fruitflies.
The Bottom Line
If we choose to take fisetin at this stage in the science, we are early adopters, and our main concern ought to be safety. There is little doubt that killing senescent cells will be beneficial. But what is the toxic burden of fisetin, and what dosage can we safely take without risk of damage to normal cells? The current study covers a lot of ground but doesn’t answer this question, apparently because they are convinced that fisetin is quite safe.
Strawberries, apples, grapes, and onions all contain fisetin, but at low levels compared to a senolytic dose. For example, the highest food concentration, 160 ppm, is found in strawberries. A half pound of strawberries yields 36 mg of fisetin. We’re still guessing at the therapeutic dose, based on mouse studies, and the experimental dosage in human trials is about 1,000 to 1,500 mg (based on this clinical trial), the content to 30-40 pounds of strawberries on each of two consecutive days.
In the best cases, fisetin was shown to reduce senescent cell burden by 50% and up to 75% in cell cultures. This is a good start, and encourages us to think we can do better by combining fisetin with other agents, or perhaps with fasting.
It sounds impressive, but I’m not impressed. First, mouse models of Alzheimer’s have been discredited repeatedly. Mice don’t naturally get AD, so they have to be genetically engineered to do so, and the genetically modified mice don’t share the deep causes of human AD. Time and again, treatments have been found effective in the mouse model that fail to translate to humans. Second, the treatment used in the study to kill senescent brain cells also relied on another genetic modification, and would not be applicable to humans.
My guess is that effective senolytic agents for humans will be available within a few years, and that they will decrease risk of all age-related disease, including Alzheimer’s. But this study does little to advance us toward that goal.
This is the most important column I’ve ever written. The message is quite complex–dozens of new health parameters to test for and to optimize, all of them interacting in ways that will require new training for MDs. The message is also as simple as it can be: There is a cure for Alzheimer’s disease. You can stop reading right here, and buy two copies of Dale Bredesen’s book, one for you and one for your doctor: The End of Alzheimer’s.
Dr Bredesen’s spectacular success is easily lost in a flood of overly-optimistic, early hype about any number of magic cures. This is an excuse for the New York Times, the Nobel Prize committee, and the mainstream of medical research, but it’s no excuse for me. I’ve known Bredesen for 14 years, and I’ve written about his work in the past. His book has been out for a year, and I should have written this column earlier.
I suspect you’re waiting for the punch line: what is Bredesen’s cure? That’s exactly what I felt when I read about his work three years ago. But there isn’t a short answer. That’s part of the frustration, but it’s also a reason that Bredesen’s paradigm may be a template for novel research approaches cancer, heart disease, and aging itself.
The Bredesen protocol consists of a battery of dozens of lab tests, combined with interviews, consideration of life style, home environment, social factors, dentistry, leaky gut, mineral imbalances, hormone imbalances, sleep and more. This leads to an individual diagnosis: Which of 36 factors known to affect APP cleavage are most important in this particular case? How can they be addressed for this individual patient?
Brain cells have on their surface a protein called APP, which is a dependence receptor. It is like a self-destruct switch whose default is in the ON position. The protein that binds to the receptor is a neurotrophin ligand, and in the absence of the neurotrophin ligand, the receptor signals the cell to die.
APP cleavage is the core process that led Bredesen down a path to his understanding of the etiology of AD 16 years ago. APP is Amyloid Precursor Protein, and it is sensitive to dozens of kinds of signals, adding up the pros and the cons to make a decision, to go down one of two paths. It can be cleaved in two, creating signal molecules that cause formation of new synapses and formation of new brain cells; or it can be cleaved in four, creating signal molecules that lead to trimming back of existing synapses, and eventually, to apoptosis, cell suicide of neurons.
In a healthy brain, these two processes are balanced so we can learn new things and we can forget what is unimportant. But in the Alzheimer’s brain, destruction (synaptoclastic) dominates creation (synaptoblastic), and the brain withers away.
On the right, one of the fragments is beta amyloid. Beta amyloid blocks the dependence receptor, so the receptor cannot receive the neurotrophin ligand that gives it permission to go on living. Beta amyloid is one of the 4 pieces, when the APP molecule goes down the branch where it is split in 4.
One of the signals that determines whether APP splits in 2 or in 4 is beta amyloid itself. This implies a positive feedback loop; beta amyloid leads to even more beta amyloid, and in the Alzhyeimer’s patient, this is a runaway process. But positive feedback loops work in both directions–a boon to Bredesen’s clinical approach. If the balance in signaling can be tipped from the right to the left pathway in the diagram above, this can lead to self-reinforcing progress in the healing direction. In the cases where Bredesen’s approach has led to stunning reversals of cognitive loss, this is the underlying mechanism that explains the success.
Amyloid has been identified with AD for decades, and for most of that time the mainstream hypothesis was that beta-amyloid plaques cause the disease. (Adherents to this view have been referred to jokingly as BAPtists.) But success in dissolving the plaques has not led to restored cognitive function. In Bredesen’s narrative, generation of large quantities of beta amyloid are a symptom of the body’s attempts to triage a dying brain.
To tip the balance back toward growing new synapses
Having identified the focal point that leads to AD, Bredesen went to work first in the lab, then in the clinic, to identify processes that tend to tip the balance one way or the other. He has compiled quite a list.
Reduce APPβ-cleavage
Reduce γ-cleavage
Reduce caspase-6 cleavage
Reduce caspase-3 cleavage
(All the above are cleavage in 4)
Reduce NF-κB (nuclear factor kappa-ligllt-chain-enhancer of activated B cells)
Increase telomere length
Reduce glial scarring
Enhance stein-cell-mediated brain repair
This explains why no single drug can have much effect on AD; it’s because the primary decision point depends on a balance among so many pro-AD (synaptoclastic) and anti-AD (synaptoblastic) signals. Addressing them all may be impractical in any given patient, so the Bredesen protocol is built around a detailed diagnostic process that identifies the factors that are most important in each individual case.
Three primary types of AD
Bredesen’s diagnosis begins with classifying each case of AD into one of three broad constellations of symptoms, with associated causes.
Type I is inflammatory. It is found more often in people with carry one or two ApoE4 alleles (a gene long associated with Alzheimer’s) and runs in families. Laboratory testing will often demonstrate an increase in C- reactive protein, in interleukin-2, tumor necrosis factor, insulin resistance and a decrease in the albumin:globulin ratio.
Type II is atrophic. It also occurs more often those who carry one or two copies of Apoε4, but occurs about a decade later. Here we do not see evidence of inflammatory markers (they may be decreased), but rather deficiencies of support for our brain synapses. These include decreased hormonal levels of thyroid, adrenal, testosterone, progesterone and/or estrogen, low levels of vitamin D and elevated homocysteine.
Type III is toxic. This occurs more often in those who carry the Apoε3 allele rather than Apoε4 so it does not tend to run in families. This type tends to affect more brain areas, which may show neuroinflammation and vascular leaks on a type of MRI called FLAIR, and associated with low zinc levels, high copper, low cortisol, high Reverse T3, elevated levels of mercury or mycotoxins or infections such as Lyme disease with its associated coinfections.
There’s also a Type 1.5, associated with diabetes and sugar toxicity, a Type IV, which is vascular dementia, and a Type V which is traumatic damage to the brain.
These categories are just a start. The patient will work closely with an expert physician to determine, first, where are the most important imbalances to address, and, second, which of the changes that cna address them are most accessible for the life style of this particular patient.
Success
Bredesen wrote a paper in 2014 about successes in reversing cognitive decline with his first ten patients. As of this writing, he has treated over 3,000 patients with the protocol called RECODE (for REversal of COgnitive DEcline), and he claims success with all of them, in the sense of measurable improvement in cognitive performance. This contrasts with the utter failure of all previous methods, which claim, at best, to slow cognitive decline.
Translation to the millions of Alzheimer’s patients will require training of local practitioners all across the country. A few doctors have already learned parts of the Bredesen protocol, and Bredesen’s website can help you find someone to guide your program, but you will probably have to travel. The first training for doctors is being organized now through the Institute for Functional Medicine.
Implications
This is a new paradigm for how to study chronic, debilitating diseases. Type 2 diabetes comes to mind as the next obvious candidate for reversal through an individualized, comprehensive program. Terry Wahls has pioneered a similar approach with MS. Cancer and heart disease may be in the future.
I’ll go out on a limb and say I think Bredesen’s protocol is the most credible generalized anti-aging program we have. (Blame me for the hyperbole, not Dr Bredesen — he has never made any such claim.) Could we adopt Bredesen’s research method to accelerate research in anti-aging medicine? Perhaps biomarkers for aging (especially methylation age) are approaching a point where they could be used as feedback for an individualized program, but Horvath’s PhenoAge clock will probably have to be 10 times more accurate to be used for individuals. Averaging over ~100 individuals can give this factor of 10 in a clinical trial. Still, we don’t have the kind of mechanistic understanding of aging that Bredesen himself developed for AD before bringing his findings to the clinic; and this is probably because causes of aging are more complex and varied than AD.
Disclaimers: I’m pre-disposed to think highly of Dale Bredesen and his ideas for 3 reasons. He was a friend to me, and gave me a platform when I was new to the field of aging. He believes that aging is programmed. And his multi-factorial approach parallels the research I have advocated for researching other aspects of aging.
I’ve been in the field of aging research from the late 1990s, just the time when Aubrey de Grey was getting his start. Before others, Aubrey had the vision to realize that cancer, heart disease, and Alzheimer’s would never be conquered without addressing their biggest risk factor: aging.
From the beginning, I admired Aubrey’s successes in communicating with scholars and the public, and I reached out to him. He has always been gracious and supportive of me personally, appreciating the large common ground that we share. There is, however, one foundational issue on which we disagreed from the start.
Aubrey regards aging as an accumulation of damage. Evolution has permitted the damage to accumulate at late ages because (as Medawar theorized in 1952) there is little or no selection against it, since almost no animals live long enough in the wild to die of old age. Aubrey’s program is called SENS, where the E stands for “engineering.” The idea is to engineer fixes to the 7 major areas where things fall apart with age.
I regard aging as a programmed process, rooted in gene expression. Just as we express growth genes when we are in the womb and ramp up the sex hormones when we reach puberty, so the process continues to a phase of self-destruction. In later life, we over-express genes for inflammation and cell suicide; we under-express genes for antioxidants, autophagy (recycling), and repair of biomolecules. I believe in an approach to anti-aging that works through the body’s signaling environment. If we can shift the molecular signals in an old person to look like the profile of a young person, then the person will become young. The body is perfectly capable of doing its own repair, and needs no engineering from us.
Over the years, research findings have accumulated, and both Aubrey and I have learned a thing or two. I’m happy to say that our favored strategies are converging, even as our philosophical underpinnings continue to differ.
A unifying idea in my research has been that aging is an evolved adaptation. This is a statement about evolutionary biology, but I came to it before I studied evolution, by looking at the phenomenology and genetics of aging.
The body does not appear to be doing its best to stay young. We can see this because when the body is under stress, it has less available resources, but manages to a better job of protecting us from aging damage. This phenomenon is called hormesis.
There are single genes that can be disabled, greatly extending lifespan in worms. Some of these have no known detrimental side-effects (pleiotropy). These could only have persisted in the genome if natural selection is favoring aging for its own sake. Similar genes exist in higher organisms, though their effects on lifespan are not as dramatic as the 10-fold increase in worms’ life expectancy in worms that comes from eliminating both copies of AGE-1.
Most genes that affect the rate of aging have been around for a long time, and do the same job. This means they are evolutionarily conserved. For example, insulin is the most effective modulator of aging in mammals (including humans). In higher animals, insulin is secreted by the pancreas, from whence it regulates blood sugar and fat storage. But yeast cells existed half a billion years before the first mammals, and have no pancreas, as my friend Barja has pointed out; and yet insulin was already a primary modulator of aging in yeast.
Programmed aging and optimism
There was a time when I spoke of “aging genes” and looked for drugs that could jam their targets and turn the genes off. Meanwhile, the science of epigenetics, or gene expression, was coming of age, so to speak. We learned that genes are turned on and off, not just in different tissues, but at different times of life. I came to think less in terms of “aging genes”, more about multipurpose genes that are deployed in appropriate combinations when we are young, keeping us strong and healthy. But as we get older, the proportions change. Aging is not accomplished via new mechanisms of self-destruction, which evolution invented for that purpose. Rather, the proportions are re-shuffled and change gradually, with effects that are more and more detrimental over time.
For example, the immune system is vital for protecting the body, but it becomes indiscriminate with age. In older people, the immune system fails to protect us from microbial infections, and simultaneously, immunity turns against the self. Autoimmunity contributes to arthritis and to Type 2 Diabetes (metabolic syndrome), as well as playing a role in AD.
For example, p53 is a gene that promotes apoptosis, or cell suicide. We need for cells to be smart enough to destroy themselves when they are infected with a virus or if they are cancerous. But later in life, apoptosis is on a hair trigger, and we lose muscle and nerve cells that are still healthy and functional.
For example, inflammation is used as a primary defense against microbes, and a way to eliminate tissue around a wound so that it can be replaced; but as we get older, signals that promote inflammation are dialed up higher and higher. Chronic inflammation contributes importantly to all the diseases of old age.
Twenty years ago, I imagined one or a few medications that would block the effects of aging genes. I wrote that the thesis of programmed aging implied great optimism about the ease with which aging might be combatted. I thought that merely lengthening telomeres might add many years to our lifespan.
Ten years ago, I saw that what was needed was re-balancing of signaling molecules to create a more youthful environment. My hope was that a few transcription factors (master regulator genes) might control a large number of signal molecules and we might set the clock by controlling just a handful of master signals.
More recently, I have come to realize that shortening telomeres are only a small part of the aging program. Worse, there is no clear line between transcription factors and hormones. Most hormones affect transcription, and most transcription factors have direct metabolic effects. There are thousands of transcription factors in the human genome. As a result, my robust optimism has been tempered, and I have come to think that we need to look for ways to re-balance a great number of genes to effect rejuvenation. I still believe in a signaling approach, but I see signals as a tangled web of cause and effect, in which every cause is also an effect, and every effect has a side-effect. Modulation of the signaling system toward a more youthful state is possible, but not easy.
Aubrey’s program, too, has changed over time
Aubrey has never believed that aging evolved as a program, but rather that aging is a manifestation of damage that is permitted to accumulate because of evolutionary neglect. Recently, he has argued explicitly against the idea of programmed aging, not for the reasons that traditional evolutionists offer, but by an argument that is uniquely his own. In his words, “it is impossible for a species to maintain two sets of genetic pathways whose selected actions diametrically oppose each other. Specifically, since we clearly have a great deal of anti-aging machinery…we cannot also have pro-aging machinery.” (My response is that we have pro-aging and anti-aging machinery that are activated at different times of life.–see Aubrey’s comment below.)
Over two decades, Aubrey, too has paid attention to research results, and his thinking about what is necessary to achieve rejuvenation is changing. I see changes in the combinations of signal molecules and call it an evolved program. Aubrey sees the same thing and calls it “dysregulation”, which is a kind of damage. Aubrey and I agree that re-balancing of hormones and other signal molecules is going to be essential.
Aubrey now finds optimism in the existence of what he calls “cross-talk”. If we engineer a fix for one kind of damage, the body may sometimes regain the ability to repair other, seemingly unrelated kinds of damage. Hence, we may not have to engineer solutions to everything—some will come for free. A dramatic example is in the benefit of senolytics. Cells become senescent over time. I see this as a programmed consequence of short telomeres; Aubrey sees it as a response to damage in the cells. But both of us were surprised and delighted to learn, a few years ago, that elimination of senescent cells in mice had 20-30% benefits for lifespan. Even though only a tiny fraction of all cells become senescent, they are a major source of cytokines (signal molecules) that promote inflammation and can cause nearby cells to become senescent in a vicious circle; this apparently accounts for the great benefit that comes from eliminating them. If we find appropriately selective senolytic agents that can eliminate senescent cells without collateral damage, then the signals that up-regulate inflammation will be cut way back, and a great deal of the work needed to repair inflammatory damage is obviated.
The SENS 7
The SENS web site still lists the same 7 categories of damage that Aubrey has used for many years. But the program to address these 7 has shifted a bit from bioengineering of exogenous solutions to signaling approaches that support the body’s innate mechanisms (which we know are sufficient to keep the body in good repair through several decades of early life). For eliminating the plaques associated with AD, SENS at one time favored the engineering of artificial antibodies that would attack them, but more recently they see promise in the discovery of Dr. Sudhir Paul that our bodies already have catalytic antibodies, each capable of destroying many antibodies and re-cycling itself for the next one. Where once Aubrey saw the need for tissue engineering to replace worn-out body parts, he now sees promise in reprogramming somatic cells to become stem cells, so that our bodies can regenerate damaged tissues endogenously. Aubrey’s 1999 dissertation in biochemistry was about the theory that aging was caused by the damage inflicted by free radicals generated in our mitochondria, but he has long since embraced the fact that free radicals have an important role as signal molecules, so that anti-oxidants are not helpful for anti-aging.
Aging is not the only threat to human life
One respect in which my thinking has always departed from Aubrey’s is that I see humans as part of a continuous web of life on earth, integrated into a global ecosystem. Aubrey doesn’t worry about the Sixth Extinction that human activity has initiated because he anticipates that future humans will invent ways to support future human life as necessary. I value nature for its own sake, and I also believe that human life depends on ecoystem support in ways for which we have seen hints, but that we have not yet begun to study. Aubrey draws a sharp line between the value of human life and the value of other life, and he is highly optimistic about the ability of our species to find new ways to sustain ourselves in a post-ecologic world.
The Bottom Line
In my youthful enthusiasm, I was entirely too optimistic about the prospects for near-term anti-aging fixes. Aubrey was probably too conservative about the scope of what needed to be done to generate man-made solutions for problems the body can’t solve itself. I have come to understand the complexity of the body’s signaling network, and the fact that it is inseparable from cellular metabolism. Aubrey has come to realize that the body has endogenous solutions that can be activated more easily than we can engineer substitutes for them. I’ve been moving the timeline out, as he has been moving the timeline in, and there is much that we agree about.
I’m grateful to Aubrey — we all are — for the energy, the expertise, and the humor that he has brought to his chosen role, as a public advocate for bringing anti-aging strategies into the mainstream of medical research.
Fo-ti is a root herb from traditional Chinese medicine that has been used for centuries as an anti-aging tonic, and has shown promise in limited Western-style analyses. Interest has been held back by reports of liver toxicity, but there is some indication that the benefits can be separated from the toxic effects. In my readings, I found anecdotal evidence for rejuvenation plus one older study of impressive life extension in quails. I also found many more recent studies documenting beneficial biochemical effects which may be counted indirect evidence that makes life extension more credible. There were two clinical trials, both with promising results.
I’ve been looking into the effects of a root herb called Fo-ti or He shou wu (何首乌) or Polygonum multiflorum Thunbergia, since a friend emailed me about rejuvenating effects when he fed it to his ancient German shepherd. I’ll call it PMT for the remainder of this page. I’ve consulted the usual PubMed sources, in addition to books on Traditional Chinese Medicine (TCM), and some scientific articles in Chinese, which I ran through Google Translate, with fair results.
TCM is based on herbal combinations and formulas. Each ingredient has many active compounds, and the art of TCM is to combine the combinations. Western medicine likes to study one compound at a time, based on a scientific tradition (reductionism) that tries to understand each separate piece, then study interactions from that understanding as a foundation. The reductionist approach was responsible for the explosive success of 19th Century physics, and has been popular ever since, but it is not obviously the best way to make progress in 21st Century biology [Carl Woese philosophy piece]. Another reason for the Western preference for single-compound treatments comes from patent law, which encourages the testing of purified compounds and disallows patents for whole plants. But our bodies are complex, homeostatic systems, and it is rarely true that the combined effect of two drugs is just the sum of the effects of each separately. Strong interactions are the rule, rather than the exception. I believe that we are not going to find a single Fountain of Youth molecule, so I have been an advocate for high-throughput screening of many combinations of treatments, looking for combinations that stand out as especially effective. If we continue to study purified molecules in isolation, it may be a long time before we get to the point where we understand the biochemistry well enough to identify magic combinations on theoretical grounds.
A curious side-note: It is reasonable to expect some combinations of biochemicals to synergize in the human body. But why should we expect these combinations to be found regularly in a single plant? Herbal medicines are unreasonably effective in this regard.
Here [1988] is the one lifespan trial that I was able to find, which reports 50% life extension in Japanese quails. I looked on Cochrane and Examine.com, and found nothing. However, Joe Cohen over at Self-Hacked has an extensive article. “More than 100 chemical compounds have been isolated from Fo-ti, and the most biologically relevant components have been determined to be from the families of stilbenes, quinones, flavonoids, and phospholipids…Fo-ti exhibits a wide spectrum of pharmacological effects, including anti-aging, immunologic, neuroprotective, anticancer and anti-inflammatory effects.” Stilbenes are molecules in the resveratrol family; quinones are like CoQ10, and flavonoids are polycyclic molecules in the quercetin family.
Most of the rest of what I report here comes from this Chinese review [王伽伯, 2016] and this English language review [Bounda & Feng, 2015].
Laboratory studies and clinical practice have demonstrated that PMT possesses various biological and therapeutic actions, including anti-tumor,[16,17] antibacterial,[18] anti-inflammatory,[13] anti-oxidant,[19,20,21] anti-HIV,[22] liver protection,[23,24] nephroprotection,[25] antidiabetic,[15,26] anti-alopecia,[27,28] and anti-atherosclerotic activities.[29,30] It has been also reported to exert preventive activity against neurodegenerative diseases,[31,32,33,34,35] cardiovascular diseases and to reduce hyperlipidemia as well.[36,37] — Bounda & Feng
Anti-inflammatory: [13] is a Korean study that found inhibition of inflammatory cytokines in white blood cells of mice. Other studies [77] show suppression of NFκB.
Liver protection: [23] A Taiwanese study that demonstrated reduced toxicity from CCl4 after mice were treated with PMT extract. [24] PMT reversed liver cirrhosis in mice.
Antidiabetic: [15] inhibits enzymes that digest starch [26] is an impressive study, that demonstrates inhibition of TGF-β1 and COX-2, and simultaneous enhancement of SOD and glutathione from a chemical extract of PMT called 2,3,5,4′-tetrahydroxystilbene-2-O-β-d-glucoside (TSG). TSG is chemically similar to resveratrol, and in a worm study was more effective than resveratrol at increasing lifespan (22%).
Anti-atherosclerotic: [29] Mice don’t get heart disease so they work with rabbits. Large reductions in measures of arterial blockage in rabbits fed a water-extract of PMT. [30] This is really about anti-inflammatory benefits of TSG fed to mice and rats.
Neurodegeneration: [31] This was about adaptogenic benefit in mice. Mice were protected from nerve damage by paraquat if they had been prepared with extract of PMT. [32] worked with a mice that had been genetically modified to give them Alzheimer’s disease. TSG was found to ameliorate the loss of memory. [33] Older rats lose their memory, as tested in their ability to remember from day to day the location of a hidden platform in a tank of water. TSG protected memory in older rats. [34] This is a study for people who believe in the Amyloid-β theory of Alzheimer’s disease. A large number of herbal substances were screened in cell lines that generate Amyloid-β, and the only effective inhibitor was found to be PMT extract. [9] Suppresses lipid peroxidation in response to Amyloid-β in a mouse model and increases glutathione. [116] Another successful trial, this time of TSG in a mouse model of AD. [119, 120 is in Chinese] Two clinical studies found substantial improvement in cognitive performance of AD patients with PMT.
Liver injury from PMT is linked to a certain genetic difference, labeled CYP1A2 * 1C. I didn’t find anything more about this genetic variant. Curiously, I found several studies that claimed that PMT protects the liver, for example this.
My inclination is to look for empirical evidence and downplay theory (both Western and Chinese theory). I believe that the emphasis on single compounds is a serious limitation of Western medical research, because the interactions are more important than the individual effects. For me, it is an attractive feature of TCM that there is so much accumulated wisdom, not just about herbs that contain many active ingredients, but about potions that combine typically a dozen or so herbs that have been found to work well together. So the maddening thing I’ve found is that the Chinese scientists who have studied PMT and other promising Chinese herbs fall into the Western trap and isolate one compound at a time to study their effects. What is missing is the lifespan studies based on whole herbs, or combinations of herbs, as they would be prescribed by a traditional Chinese herbalist.
I went to a local herbalist this week and asked for advice about He Shou Wu. She explained to me that in TCM, herbs are always given in combinations. There are classic formulas with 6 or 10 or 20 herbs, and these are adjusted for individual prescription. The main ingredients are large quantities of the herbs that move the metabolism in some direction, and the lesser ingredients counterbalance the main ingredients by pushing in the opposite direction. Some of the directions they talk about correspond to observables we might recognize (high or low energy, sexual stimulant), and some of them are more esoteric (wet or dry, hot or cold, yin or yang). She gave me a formula with He Shou Wu as the main ingredient, and I’m going to do some more reading before I decide whether to take it.
In the meantime, I’m taking a gram of He Shou Wu extract processed with black beans each morning before breakfast and I think I detect an increase in aerobic stamina which has not listed anywhere as one of the benefits.
Acknowledgement: The idea for this research came from Jeff Bowles, who is a frequent commenter on this blog. The Chinese research was kindly supplied by Wen-jun Li, a post-doc in the Beijing lab where I have worked the last 3 summers.
This week, a headline-making study in the New England Journal of Medicine sought to cast doubts on long-established science that says daily aspirin can be a broadly-effective anti-aging tonic. I’m writing this response because I think that this new, small study has to be viewed in the context of many larger studies over many decades that together make a solid case for aspirin’s benefits.
Aspirin has two kinds of effects: First, aspirin thins the blood, reduce clotting, which lowers the risk of most kinds of heart attacks and stroke (ischemic) while raising the risk of bleeding ulcers and hemorrhagic stroke. Second, aspirin lowers the level of systemic inflammation, which reduces risk of heart disease, stroke, most cancers, and Alzheimer’s disease.
Historically, daily low-dose aspirin began to be prescribed broadly to middle-aged and older adults in the 1960s as the medical establishment theorized about the first effect. This led to a grand natural experiment—tens of millions of older people taking low-dose aspirin. Studies comparing these people with matched populations who didn’t take aspirin have shown lower rates of all-cause mortality, Alzheimer’s dementia, and of cancer and probably of heart disease as well. These studies are based on millions of tabulated deaths. The current study is based on 1052 total deaths in the aspirin group and the placebo group, and the difference between the two was barely statistically significant in the direction against aspirin.
Summary of past studies
Eidelman, JAMA, 2003: Summarizing 5 trials, they found aspirin was associated with a 32% reduction in the incidence of first heart attacks. Statistical significance was 2 chances in 100,000 (p<0.00002).
MethodsA computerized search of the English literature from 1988 to the present revealed 5 published trials: the Physicians’ Health Study (22,071 participants), the British Doctors’ Trial (5,139), the Thrombosis Prevention Trial (5,085), the Hypertension Optimal Treatment Study (18,790), and the Primary Prevention Project (4,495).
Results Among the 55,580 randomized participants (11,466 women), aspirin was associated with a statistically significant 32% reduction in the risk of a first MI and a significant 15% reduction in the risk of all important vascular events, but had no significant effects on nonfatal stroke or vascular death.
ConclusionsThe current totality of evidence provides strong support for the initial finding from the Physicians’ Health Study that aspirin reduces the risk of a first MI. For apparently healthy individuals whose 10-year risk of a first coronary event is 10% or greater, according to the US Preventive Services Task Force and the American Heart Association, the benefits of long-term aspirin therapy are likely to outweigh any risks.
Rothwell, The Lancet 2011: Summarizing 8 trials, they found aspirin was associated with a 21% reduction in the incidence of all cancers. Statistical significance was 1 chances in 10,000 (p<0.0001).
Results
In eight eligible trials (25,570 patients, 674 cancer deaths), allocation to aspirin reduced death due to cancer (pooled odds ratio [OR] 0·79, 95% CI 0·68–0·92, p=0·003). On analysis of individual patient data, which were available from seven trials (23,535 patients, 657 cancer deaths), benefit was apparent only after 5 years’ follow-up (all cancers, hazard ratio [HR] 0·66, 0·50–0·87; gastrointestinal cancers, 0·46, 0·27–0·77; both p=0·003). The 20-year risk of cancer death (1634 deaths in 12 659 patients in three trials) remained lower in the aspirin groups than in the control groups (all solid cancers, HR 0·80, 0·72–0·88, p<0·0001; gastrointestinal cancers, 0·65, 0·54–0·78, p<0·0001), and benefit increased (interaction p=0·01) with scheduled duration of trial treatment (≥7·5 years: all solid cancers, 0·69, 0·54–0·88, p=0·003; gastrointestinal cancers, 0·41, 0·26–0·66, p=0·0001). The latent period before an effect on deaths was about 5 years for oesophageal, pancreatic, brain, and lung cancer, but was more delayed for stomach, colorectal, and prostate cancer. For lung and oesophageal cancer, benefit was confined to adenocarcinomas, and the overall effect on 20-year risk of cancer death was greatest for adenocarcinomas (HR 0·66, 0·56–0·77, p<0·0001). Benefit was unrelated to aspirin dose (75 mg upwards), sex, or smoking, but increased with age—the absolute reduction in 20-year risk of cancer death reaching 7·08% (2·42–11·74) at age 65 years and older.
Wang, Journal of Alzheimer’s 2015: Summarizing 11 trials, they found aspirin was associated with a 49% reduction in the incidence of dementia. Statistical significance was less than 1 chances in a billion (p<0.0000000005).
Abstract
Objective: Alzheimer’s disease, the most prevalent dementia, is a prominent source of chronic illness in the elderly. Laboratory evidence suggests that nonsteroidal anti-inflammatory drugs (NSAIDs) might prevent the onset of Alzheimer’s disease. Since the early 1990s, numerous observational epidemiological studies have also investigated this possibility. The purpose of this meta-analysis is to summarize and evaluate available evidence regarding exposure to nonaspirin NSAIDs and risk of Alzheimer’s disease using meta-analyses of published studies. Methods: A systematic search was conducted using Medline, Biological Abstracts, and the Cochrane Library for publications from 1960 onwards. All cross-sectional, retrospective, or prospective observational studies of Alzheimer’s disease in relation to NSAID exposure were included in the analysis. At least 2 of 4 independent reviewers characterized each study by source of data and design, including method of classifying exposure and outcome, and evaluated the studies for eligibility. Discrepancies were resolved by consensus of all 4 reviewers. Results: Of 38 publications, 11 met the qualitative criteria for inclusion in the meta-analysis. For the 3 case-control and 4 cross-sectional studies, the combined risk estimate for development of Alzheimer’s disease was 0.51 (95% CI = 0.40–0.66) for NSAID exposure. In the prospective studies, the estimate was 0.74 (95% CI = 0.62–0.89) for the 4 studies reporting lifetime NSAID exposure and it was 0.42 (95% CI = 0.26–0.66) for the 3 studies reporting a duration of use of 2 or more years. Conclusions: Based on analysis of prospective and nonprospective studies, NSAID exposure was associated with decreased risk of Alzheimer’s disease. An issue that requires further exploration in future trials or observational studies is the temporal relationship between NSAID exposure and protection against Alzheimer’s disease.
Problems with the present study
Because of small numbers and short duration, the result of the study was only marginally significant (p<0.05). The aspirin group had higher cancer rates and lower heart attack rates than placebo.
Typically, doctors advise patients to start low-dose aspirin around age 50, but this study was with patients more than 70 years old who had no cardiovascular symptoms by age 70. Most people by age 70 have had some cardiovascular diagnosis before age 70, so this is an unrepresentative sample. The study fails to address the question, how many deaths and how many diseases could be avoided between the ages of 50 and 70? This is the period in life when inflammation is most active, and a great deal of destruction of the body’s veins, joints, and nervous system happens during these years. Excluding those with a history of heart disease during those ages is excluding just the people most likely to be helped by aspirin. Of course, when you’re 50 and considering whether to start on aspirin, you may not know whether you’re lucky enough (or have the right genes) to be in the group that will do fine for the next 20 years without it.
This table breaks the composite test group into sub-groups according to various criteria. Dots to the right of the line mean “aspirin was worse”, and to the left mean “aspirin was better”. Among the subgroup in the US, aspirin was better. Among people who had never taken aspirin before, aspirin was better. Among people within fairly wide limits of a “normal” weight range, aspirin was better.
Why are we seeing this?
Scientists are only human, and their environment, their preconceptions, and their incentives shape the way that statistics are handled. In my experience, it is not difficult to make a small effect look like a (p<0.05) effect by making consistent choices in the way the data are treated, none of which are suspect or dishonest. If the group had come up with the conventional and accepted conclusion based on such a small study, there would have been no prominent publication, no headlines, probably no follow-on grant. So they had every incentive to perform the analysis in a way that makes the results appear more interesting than they are.
[My sources for much of this article are a 2018 review from University of Campinas, Brazil and a 2016 review on hormesis by Joan Smith Sonneborn, as well as the ever-inspiring and accessible summaries by Rhonda Patrick.]
Most animals have the latent ability to live longer when stressed. It’s called hormesis, and it’s a major clue concerning the nature and evolutionary provenance of aging. The body compensates when stressed—that’s no surprise—but the remarkable thing is that it overcompensates so that, paradoxically, stress ends up by lengthening lifespan. Sometimes.
One of the prime responses to stress at the cellular level is Heat Shock Proteins, discovered in 1962 in fruit flies. Heat was the stressor that led to the original discovery of HSP, and the word “heat” remained with the name, though it soon became clear that HSP are secreted in response to many kinds of stress, including cold. HSP are not a single protein, but a family of molecules, all of which are highly conserved; the human versions are remarkably similar to HSP in flies and even yeast cells.
HSP protects delicate biomolecules from damage. HSP act as chaperones, helping newly-created proteins to fold properly, and helping misfolded proteins to find their correct shape. HSP protect against sarcopenia (muscle-wasting) which is responsible for so much frailty. Lab worms with an extra copy of an HSP gene live longer. Here is a closely-related finding for fruitflies, but there are contradictory findings for mice [pro, con].
Heat Shock Factor (HSF) is a signal molecule that turns on the full set of HSP genes. It turns on a great many other protective proteins at the same time, a whole library, in fact, of protections. Calorie restriction and exercise both activate HSP, but protein restriction may attenuate HSP. HSP induction in response to HSF declines with age in rodents, but not if they are calorically restricted. Pro-biotics and high-fiber diets encourage microbiome signaling that increase HSP expression, at least in mice. Insulin resistance, characteristic of type 2 diabetes, suppresses HSP in response to HSF. High fat diets reduce HSP. Garlic in the diet increases HSP.
HSP is neuroprotective when there is potential damage from a stroke or head injury. Does HSP protect nerves from the slow damage of aging as well?
Saunas
In my reading this week, I’ve come to think that saunas may be the second most powerful form of human hormesis after calorie restriction. Statistics for saunas suppressing cardiovascular disease and especially dementia make you stand up and take notice. Here’s a clear and straightforward article by Rhonda Patrick (FoundMyFitness) about the benefits of saunas.
If you’ve ever run long distances or exercised for endurance, it’s intuitive that increased body temperature will eventually induce strain, attenuate your endurance performance, and accelerating exhaustion. What might not be as intuitive is this: acclimating yourself to heat independent of aerobic physical activity through sauna use induces adaptations that reduce the later strain of your primary aerobic activity. Hyperthermic conditioning improves your performance during endurance training activities by causing adaptations, such as improved cardiovascular and thermoregulatory mechanisms.
I don’t enjoy getting overheated any more than you do, but hey—stress is stressful. How surprised can we be that heat is a powerful inducer of Heat Shock Protein? Perhaps more interesting is that saunas are associated with increased growth hormone, a far safer and cheaper way to achieve higher HGH levels than injections. The combination of HGH and HSP help to maintain muscle mass against the erosion that almost always comes with age. Patrick documents that saunas contribute to maintaining (or restoring) insulin sensitivity, and to growth of new brain cells. Another pathway by which saunas work their magic is norepinephrine=noradrenaline, which is both a neurotransmitter and a hormone, and higher levels are associated with good attention and cognitive performance.
“The greater the discomfort experienced during your workout or sauna, the better the endorphin high will be afterward.”
Jari Laukkanen, a Finnish cardiologist, followed middle-aged sauna-bathers (men) and matched controls for 20 years. His study found dramatic decreases in cardiovascular deaths, and a 40% drop in all-cause mortality for those reporting sauna use at least 4 times per week for 20 minutes. A prospective study–planned in advance to follow 2,300 men over 20 years–is the gold standard for epideiology. A 40% drop in mortality is worth about 3 years of extended life. An even more impressive number: the Alzheimer’s risk of men taking at least 4 saunas a week was only ⅓ as great as those who took 1 sauna a week. The benefit compared to no saunas at all is likely to be substantially greater yet.
Just this week, there is a new review by Laukkanen, author of the above study, who also did much of the the original research in his review.
The review doesn’t mention cancer, and there have been mixed reports whether saunas and HSP in particular protect against cancer or add to cancer risk. On the one hand, localized applicatation of heat and even whole body heat are a well-established cancer treatment over 40 years. On the other hand, HSP increases the ability of cells to survive stress, and that includes cancer cells. There is some evidence that saunas enhance the immune system and that would likely contribute to cancer resistance. In my judgment, the balance of the evidence is that saunas lower cancer risk.
Choose your poison.
The body responds to alcohol as a poison, and raises levels of HSP. This may be the mechanism by which alcohol consumption (~1 drink per day) lowers heart attack risk, though cancer risk is increased even at low doses.
I’ve made my choice, and I’ve been a teatotaler my whole life. It’s been for personal reasons that I never have written about the established epidemiology of alcohol. Moderate alcohol consumption has conventionally been associated with a modest increase in life expectancy, (~1 year or less), but conventional wisdom could be wrong. It’s always difficult to separate variables in large population studies, and alcohol consumption is linked to so many different factors, all of them more powerful influences than alcohol itself.
Cold
HSP is a stress adaptation, not specialized to heat, and in fact cold temperature can also trigger release of HSP. That said, cold and heat are not symmetric. Saunas work by raising the core temperature of the body several degrees, as in a fever. Cold is applied on the skin, and the core of the body works harder to keep its temperature close to normal. The benefit is mediated by the cold-sensing nerves in the skin, which trigger release of norepinephrine, similar to heat exposure. A specific response to cold is a protein called RMB3, which promotes neurogenesis.
It’s tempting to take your cold shower or plunge into an icy stream after you’ve been working out and your core temperature is elevated. But this may actually cause delayed cramping and lessen the benefit of your workout. I hate to say it, but after resistance training is the most beneficial time to take your sauna (if the least comfortable). If you can’t bear the thought of jumping into a cold shower when your body is already cold, you might try a hot shower first. Here’s a study that demonstrates a drop in infectious disease rates from hot showers followed by cold. Hof recommends that you take your cold plunge after a course of deep breathing.
One of the most consistent and profound physiological responses to cold exposure is a robust release of norepinephrine into the bloodstream, as well as in the locus coeruleus region of the brain. — Rhonda Patrick
Does the Wim Hof method increase life expectancy
In the last several years, Dutch extreme athlete Wim Hof has popularized a training discipline that combines breathing exercises, cold immersion, yoga and meditation.
Wim Hof is able to suppress immune response to a standard challenge, suggesting he is also able to consciously suppress the auto-immune response that contributes to arthritis, and probably diabetes and AD as well. When Hof was studied with metabolic and neurologic sensors, the result indicated that he has acquired conscious control over physiological adaptations which, in the rest of us, are entirely automatic. Is it possible to learn to dial down inflammation by an act of will, or to control our epigenetic age directly from the mind? This is an approach to health and perhaps to anti-aging that has always fascinated me, though there is little in the mainstream literature on the subject because it is presumed impossible. There have long been stories about yogis and ascetic devotees of Eastern religions who culture extraordinary control over their bodies and live to extraordinary ages. Of course, we would like to see these claims subjected to controlled conditions and standardized lab tests, but there are probably good reasons why most ascetic hermits have no interest in taking leave from their mountain caves to serve as lab rats.
There is no direct evidence that Wim Hof training affects aging. Indirect evidence is that it lowers inflammation, which makes a large contribution to all the diseases of old age, and that it releases norepinephrine and RMB3, both of which are neuroprotective I’m eager to see if Wim Hof method has an effect on methylation age, and will include it in the Data BETA study that is ramping up this fall (DataBETA is the name I’ve chosen for the Mother of All Clinical Trials. It stands for Database for Epigenetic Evaluation of Treatments for Aging.)
The Bottom Line
If the Finnish review is to be believed, then hyperthermia—overheating—is one of the most powerful modes of hormesis we know of, ranking second only to calorie restriction. Just as interesting is the fact that hyperthermia works by a path independent of insulin, so we might hope that there is synergy between saunas (or Bikram yoga) and from calorie restriction (or fasting). In other words, combining low calorie with high heat might, if we’re lucky, yield life extension equivalent to the sum of the two measures separately. Cold exposure and the full Wim Hof program, including meditation techniques, show promise, but are further from validation as a life-extending practice.
What are the most effective things you can do to slow the aging process and extend your life expectancy? This is the question being asked by a clinical trial that I am organizing, and which seems to be rapidly taking shape. But before the study begins, we have to have candidates to evaluate. We should begin with hypotheses about what we are evaluating. My idea is to consult some experienced experts, and also to crowd-source this choice, and to ask for your help in selecting the supplements and life habits to be evaluated.
Details of the trial were described in two blog posts last spring [One, Two] and a more technical manuscript submitted in May. Outcome will be evaluated based on a variant of DNA PhenoAge, taken from a blood test before, amid, and after the two-year trial. We use methylation pattern differences rather than mortality or health outcomes because the latter take a long time to reveal themselves, and make anti-aging trials prohibitively expensive. Using methylation clocks as an endpoint is a new idea, and we don’t know if it will work, but if it does, it will be 100 times cheaper and 10 times faster than previous methods. We will have enough bandwidth to test a dozen different measures at once, which itself is a revolutionary step.
Many measures are known that are thought to increase life expectancy by a year or a few years each. Of course, we want to know which ones offer their greatest benefits. But even more important, we want to know how they interact, synergize, and interfere with one another. If any one of these measures offered major benefits—say 20 years of life—its effects would be so apparent that we would probably know it already. Likewise, if these measures added up to 20 years of extra life, we would all know some people who are obviously younger than their chronological age. Realistically, we must assume that most of the things we do are redundant. Combining metformin with berberine and gynostemma may offer little additional life expectancy compared to any one separately. A panoply of different anti-inflammatory strategies may be little improvement over an aspirin a day.
But we hope there are exceptions. If two different measures act via completely different metabolic pathways, we have reason to hope that their effects should compound. For example, perhaps life extension interventions based on mitochondrial health synergize with interventions based on rebooting the immune system. Then we might hope that the life extension available from these two measures together is greater than the sum of what we get from the two separately.
Study design
We will not tell the people who sign up for this study what to eat, what pills to take, or how much to exercise. We will ask people what they are doing, what they are eating, and what supplements they are taking in a detailed questionnaire. We will select subjects so as to represent a broad array of different strategies and different combinations among these strategies.
Broad, but not too broad. We will have enough statistical power to estimate the interactions among every pair of measures out of 12 that we take as our independent variables. These 12 should be chosen in advance, so the study has a clear focus. If there are more than 12, the number of interactions increases rapidly beyond what we can hope to distinguish (with multivariate statistics). I’ve decided to start with 15, and winnow the list as people sign up for the study and we see what
Here are the criteria I propose:
Each measure, separately, should have either human mortality data or longevity data to back it up.
The measures should be easily available to all (excluding intravenous drugs or transfusions)
The measures should be well-enough known that they are already in common use (and we will have no trouble identifying a diverse group of subjects who use them)
I find that it’s hard to limit the list to 15. It may make sense to include a few measures that are so well established that every participant will be required to comply in order to be included in the study. In this category, we might put
Non-smoking
Limited alcohol consumption (or none)
Vitamin D at least 5,000 IU daily
Multi-mineral supplement with magnesium, zinc, chromium, and selenium
Exercise equivalent to minimum 5 hours a week of walking or yoga
Body weight (BMI) is an important longevity factor, but difficult to account. Studies show that maximum lifespan is associated with BMI between 21 and 25, but in my interpretation, lower BMI is always beneficial for any given individual.
The reason for the apparent paradox is that individuals have genetic disposition to be overweight or underweight. Those who are genetically underweight tend to overeat, because they can do so with no social stigma. Those who are genetically overweight feel compelled to diet all the time (women more than men), and they may be restricting calories just to keep their BMI at 25. These dieters get the most benefit from Caloric Restriction, despite the fact that they don’t look thin.
List of things that lengthen your life
Love. Men who are married or in close relationships have 7% lower mortality than singles. The number is 4% for women [ref]. These numbers correspond to less than a year of life expectancy. A different study finds loneliness increases mortality by 50%, corresponding to almost 5 years of life.
Empowerment: Staying employed is worth up to 14 years, and I like to think this is more about being needed than making money. This study claims that the big difference is wealth.
Anti-inflammatories: Aspirin, ibuprofen, curcumin, or fish oil. This study attributes about a year of life expectancy to daily aspirin.
High fiber, a proxy for healthy gut flora. We know that they are important, but don’t yet know how to manage the biota for maximal life expectancy.
Vegetable-based diet This review concluded that vegetarians live 3 years longer, but methodologies and results vary considerably.
Meditation This is difficult to evaluate, and data is unreliable but encouraging [ref]. The only careful study linked meditation not to mortality but to telomerase activity. I confess I am including meditation in the list from my own intuition and experience.
Intermittent fasting Extends lifespan in mice and lowers mortality in nursing home studies [ref].
Interval trainingReputed to be the most efficient path toward cardiovascular fitness [ref], and there is limited documentation of benefit for all-cause mortality [ref].
Donating blood This is another quirky inclusion on my part, but there is data to support it, which I reviewed a few years ago.
NAC just one study — 30% increase in lifespan of mice
DHEA Lower blood levels of DHEA are clearly associated with greater age and higher mortality at the same age, but the direction of causality is in dispute.
Metformin Prescribed for diabetes for decades, this drug also lowers mortality from cancer and heart disease [planned clinical trial].
RapamycinThe most convincing data available for any supplement for life extension in mice. Early adopters are beginning to experiment on their own.
Quercetin + Dasatinib (or other senolytics). Senolytics are the best near-term hope we have for a breakthrough in anti-aging medicine; but the combination of quercetin +dasatinib is not yet discriminating enough to be safe for humans, meaning it kills too many regular cells.
Epithalamin Very promising data from Russia [ref], both in rodents and in people, but there is no one trying to reproduce them in the West.
Selegiline (deprenyl). In classic studies from the 1980s, lifespan of rats was extended. Data in humans is contradictory [ref].
Perhaps we should begin with a guess about which combinations are likely to be highly redundant. In this way, we could cluster together different strategies and condense more strategies into a manageable list. It might look like this:
Or—the best of both worlds—we might structure the study in such a way that it can be analyzed after the fact either with individual strategies or clusters of strategies.
Fauja Singh, centenarian marathon runner
The best way to design a study is to start at the end. I imagine I am two years down the road, taking my first look at results from 5,000 life-extenders. The first thing I will want to do is to look for outliers. Are there a few people who stand out from the bell curve, aging much more slowly? If so, what do they have in common? The advantage of this approach is that it gives maximal flexibility in telling us exactly what we most want to know. The disadvantage is that it is easy to fool ourselves and imagine patterns in a small set of random errors. When there is no initial hypothesis, there is no objective way to calculate a probability that what we find is the result of chance.
Our null hypothesis is that there are no systematic outliers, but only a smooth tail to the probability curve. If there are a few scattered individuals in 5,000 who are aging much more slowly than the rest, we will not find any common thread in what they are doing, and so we must explain their data as anomalies or mistakes. If this is our result, it will be disappointing, sobering, but liberating as well. Those of us who are compulsive about one or another life extension strategy can ease our discipline.
But there is a chance we will find something more interesting. We may find that there are dozens of outliers, that their life extension strategies all overlap in some clear and unambiguous way. We will then have, for the first time, a solid foundation for our personal life-extension habits, and a clear hypothesis for further experiments.
The Mother of All Clinical Trials, which I announced in April, continues to progress at a charmed pace. This is a project to collect information from people who are already using a variety of measures to extend their life expectancy, and to use a methylation clock and some innovative statistics to tell us which combinations are effective. It is to be an open-source study, with all data, results and analysis freely available to the public and the research community.We have done no fundrasing as yet, but have collected remarkable volunteer talent and a mammoth donation-in-kind from Zymo Research, the only company that presently offers methylation age testing commercially.
Our principal unfilled role is a project director, who will recruit, train and manage 5,000 subjects from within the life extension community, oversee collection of data, and keep them motivated. This could be a volunteer position, or it may involve fundraising your salaray + related expenses.
We are also looking for a lawyer who can advise us on privacy, HIPAA, IRB matters, and related IP issues. Write to Josh Mitteldorf <[email protected]> if you are interested in working with us.</[email protected]>
My Himalayan Experience
I’m going to invoke my prerogative as a blogger and talk a bit about my personal experience. Two weeks ago, I was trekking in the Himalayas. This adventure has been on my bucket list for many years, both because of the grandeur of the landscape and the challenge of exercise at high altitudes. This spring, I finally got around to it.
your blogger, day 3
On the rare occasions when I’ve been above 4,000 meters in the past, I was a little short of breath but didn’t have headaches or nausea that are commonly experienced. A week before my trip, I looked up the Chinese word for altitude sickness and stopped into the herbal pharmacy at the shopping mall near where I was living in Beijing. The pharmacist offered a box of pills, whose ingredients included rhodiola, Goji berry, ginseng and taurine, all of which have some evidence as longevity aids.
I arrived at Lhasa airport at 7 in the evening (3600 meters) but the sun was still high in the sky, due partially to the fact that it was close to summer solstice, but mostly because China’s single time zone should really be 3, and Tibet is in the far west. I had a limo to the city, and didn’t feel bad at all. I went out for a late dinner, and felt the first headache symptoms as I went to bed. In the middle of the night I awoke with a rip-roaring headache, and a sense of déjà vu. Only then did it dawn on me that I had forgotten to ask the waiter at my restaurant to avoid MSG in my meal, a mistake which I had already made twice before in my 8 weeks in Beijing. The headache was gone by mid-morning, and never returned during my week in the Himalayas.
For the trek, I was tacked onto a group from Singapore, all half my age. We were out at 4,000 to 5,000 meters over four days, covering about 20 Km per day of ups and downs. Air at 5,000 meters is just about half the pressure (half the O2) compared to sea level. I never felt sick, but I was out of breath whenever we walked uphill, even a small incline. For the second day out when we first crossed 5,000 meters, I was doing kapalabhati for hours on end (fast, yogic belly breath) — pumping air into my lungs as fast as I could to avoid the lightheadedness that would stop me in my treks.
Apnea – the mind cure
I have had sleep apnea for 20 years. When I’m asleep, my body forgets to breathe, until my brain senses oxygen deprivation, startles me half-awake, I gasp a few breaths, fall back asleep, and the same cycle repeats. I’ve just barely managed the condition by sleeping on my stomach. Sometimes I’m aware of the apnea as it happens, but mostly I’m not; during the day I have bouts of sleepiness, presumably because my nighttime sleep is not deep, and I’m fortunate that usually I have the freedom to take naps as needed.
What I didn’t learn until I got to Lhasa: Altitude makes apnea worse. On the one hand, there’s less oxygen, so we need to be breathing faster; on the other hand, there’s also less CO2, and it’s the buildup of CO2 in the blood that the body senses in order to regulate breathing. I usually take 1 mg melatonin at bedtime, for longevity benefit rather than for sleep. While in Tibet, I suspended melatonin because statistically it exacerbates apnea, and in my experience, melatonin at higher doses seemed to be a major factor.
My first night out on the trail, I really sensed the apnea, much more so than in Lhasa. I repeatedly felt myself startled awake, panicked and panting. I wasn’t sleeping much.
The second night, my difficulty sleeping was more severe, and I was inspired in the middle of the night to try an experiment. I sat erect in a meditation pose and found a rhythm that gave me enough air = 3 heartbeats inhale, 5 heartbeats exhale. (This was about three times faster than my resting breath at home.) I used meditation techniques to keep my mind returning to the breath, aware of the rhythm, and aware when the O2 budget felt insufficient, and I needed to breathe deeper and faster for a bit. After about 20 minutes, I lay down and maintained the same counts, the same breathing rhythm, the same relaxed, meditative mental posture. I deliberately formed the intention to impress the rhythm on my unconscious, so that it might continue to breathe in the same pattern after I dozed off. The technique worked. It was awhile before I dozed off, but the time meditating was fully relaxing, and gave me the feeling that my brain and body were restoring as they might have if asleep. When, eventually, I did fall off to sleep, there was no panicked awakening. I can’t be sure whether the apnea was returning because I was in a tent alone, but as far as I could tell, it was relaxing sleep.
I regard the experience as a breakthrough in my relationship with apnea, and I’ve continued to rhythmically breathe myself to sleep in the 2 weeks since I’ve returned to sea level.
Adaptation to Altitude
Many peoples around the world who are adapted to high altitude living have more red blood cells. This works to carry more oxygen more efficiently to the tissues, but high RBC inclines the blood to clotting, and increases risk of heart disease and stroke. The Himalayan peoples have a better idea. They actually have lower RBC counts than the rest of us, but they have a genetic variant known as EPAS1 that enable their mitochondria to function just fine, to burn sugar efficiently at low oxygen levels.
Until recently, the origin of EPAS1 was a mystery. Then, in 2014, the geneticists traced it to a group called the Denisovans, 40,000 years ago. Denisovans were an offshoot of Neanderthal man, chronicled from a single finger bone of a single young woman, found in a cave in Siberia in 2010. The bone had enough DNA to do a complete sequence, and an entire subspecies known fro this single example. The Denisovans interbred with other human tribes of Asia, and the EPAS1 gene was originally their contribution to humanity. It disappeared in many places, but in Tibet, it was useful, so it stuck.
It may be counterintuitive that more is not better when it comes to red blood cells. P.D. Mangan has been beating the drum to advise us that iron levels on the low side of normal are better not just for cardiovascular risk, but for many other aspects of health as well.
Benefits of Hypoxia
Tibetans have short life expectancy compared to other Chinese groups. This may be due to poverty and inadequate access to medical care. But, curiously, there is also a high concentration of nonagenarians and centennarians in Tibet. Could altitude be a factor?
There is indirect evidence linking hypoxia to longevity. Hypoxia shifts gene expression toward a stress response that is known to overlap with longevity genes [ref, ref]. Hypoxia increases lifespan in bees [ref], fruitflies [ref], and lab worms [ref]. A study correlating altitude with life expectancy across the US found tentative evidence for a benefit from living at higher altitude.
I’m not impressed by the arguments that hypoxia is a factor in the longevity of whales, naked mole rats, and other animals whose lifestyles incidentally lead to hypoxia–too many confounding variables.
Evidence on apnea
Apnea is two separate diseases. Obstructive Sleep Apnea (OSA) has a mechanical origin in blockage of the windpipe. It is associated with obesity, but studies find that independent of obesity, apnea is a mortality risk. Central Sleep Apnea (CSA) originates in the central nervous system, but its logic and mechanisms remain obscure.
OSA incidence increases modestly with age. CSA increases dramatically with age.
CSA is much rarer than OSA, but its incidence increases dramatically after age 65. (For CSA, I was unable to find a graph like the above.) CSA is associated with heart disease and stroke, and the direction of causality is unclear. It may be both that heart failure contributes to apnea and apnea contributes to heart failure [ref]. For those of us who suffer from CSA, it would be interesting to know if treating the symptoms (say, with CPAP) lowers cardiovascular risk. Consensus of the medical community is “yes”, but this conclusion may be driven by economic and legal factors. I have been unable to find a definitive answer in the primary literature, because the direction of causality is so hard to discern. This small study (2005) found a major decrease in 5-year CV mortality for those who accepted CPAP treatment compared to those who could not tolerate CPAP. This larger study (2016) found that CPAP effectively alleviated the symptoms of apnea, but had no discernible effect on CV mortality. Of course, better sleep at night and better alertness during the day are sufficient reasons to treat the symptoms of apnea. But some of us aren’t helped by CPAP.
The Bottom Line
Hiking at high altitudes is a great challenge, but not necessarily the best conditioning for long life. Unless you’ve got Denisovan genes, you will adapt with higher red blood counts, which, for most of us, is a net negative.
Sleep apnea is entwined with heart disease, so it is difficult to separate cause and effect. Lowering the risk factors for apnea may be as important as treating the apnea itself. There is but little indication that sleep quality directly affects your mortality risk, but it certainly affects quality of life.
From what I have seen, there is a well-established correlation between apnea and increased mortality, especially CV mortality, but it is not clear that apnea patients using CPAP have lower mortality than untreated apnea patients. I’m taking a controversial position based on 2 days’ reading, and I could be very wrong about this, so I invite response and discussion.
My own experience suggests that it’s possible to use meditation techniques to plant suggestions in the unconscious that alleviate sleep apnea and improve sleep quality. Hypnotism, autosuggestion, and biofeedback might be effective as well. It’s hard to do controlled studies to demonstrate this benefit, and it may be even harder to get them funded. But it’s an approach worth exploring.
Time and again, evolution has learned (after repeated blind alleys) to do what is best for the community in the long term and not always what is best for the individuals in the short term. But such gains are fragile, easily lost if a cheater can gain a short-term advantage and its progeny take over the community.
Human societies have rules that encourage cooperation, and enforcement mechanisms for people who are reluctant to cooperate. Cooperation in biology is very old, and it turns out that evolution thought about enforcement a billion years before Thomas Hobbes. To see what this has to do with theories of aging, you’ll have to be patient.
Story #1: Conjugation and Cell Senescence
Story #2: Sex Required for Reproduction in Plants and Animals
Story #3: Antagonistic Pleiotropy — a Revisionist Theory
To begin, I’m going to ask you to think fresh thoughts about sex. (Have I lost you already?)
Sex and reproduction, reproduction and sex. Go together like a horse and carriage, right? Well, how did it come to be that way? Sex is not a way to reproduce. Sex is a way to share genes. But sex has become so tightly linked to reproduction that it requires mental gymnastics to imagine that it might have been otherwise.
Reproduction without sex—that’s not too hard. It’s cloning. Or it’s mitosis, simple cell division which is how bacteria do it.
But sex without reproduction? What’s that? Remember—sex is the mixing of genomes between different individuals with different genomes. Does anyone do that except as a prelude to reproduction? What would it even look like?
Bacteria share genes willy-nilly. They shed plasmids, which are little loops of DNA, and they pick up plasmids from around them. The plasmid may be from the same kind of bacteria or another kind of bacteria entirely. Sometimes the gene they pick up is useful; sometimes, not so much; sometimes the imported gene kills them. Bacteria can afford this daredevil lifestyle because there are a lot of them, and their credo is experimentation. Change or die. Bacteria are constantly changing, not only because their generations are measured in hours instead of years, but the change from generation to generation is also greater than large animals and plants. Under stress, they mutate and change even faster. Bacteria are artists of change, and their genius is figuring out what it takes to survive in the environment where they happen to be now. For bacteria, sex is spitting out plasmids and picking them up.
Bacterial plasmid (electron micrograph)
Story #1: Conjugation and Cell Senescence
Protists, or protoctista, are single-cell eukaryotes—far more complex and structured than bacteria, with a cell nucleus and many more organelles, a million times bigger than bacteria but still a single cell. Examples are amoebas and paramecia. Protists share genes by a process called conjugation that challenges our idea of the individual. As promised, sex in protists is not linked to reproduction…well, maybe indirectly linked, as we’ll see.
(This movie isn’t conjugation; it’s a hunting expedition.)
In conjugation, two paramecia (Dick and Jane) sidle up to each other and their cell membranes coalesce, forming one big cell. Then the cell nuclei, where the chromosomes live, find each other and the two nuclear membranes open up and merge, just as the cells did. A double size cell with double size nucleus, and two copies of each chromosome. Somehow the chromosomes pair up with the appropriate partner. Like blind people trying to navigate a crowded room, how do the chromosomes arrange a meeting place with their partners? (If chromosomes had telephones, I suppose they would be cell phones. OK, it isn’t funny.) Somehow, Dick’s chromosomes finds Jane’s corresponding chromosome, nearly identical but for the crucial variations that make them individuals. The chromosomes line up in pairs so they can swap genes with one another. Genes cross over until each chromosome contains about half Dick’s genes and half Jane’s. Then—again using their cell phones for coordination—the chromosomes segregate. One from each pair goes north, the other goes south, so that when the nucleus splits in two again, each half has a full complement. Two cells go their separate ways, but the cells that emerge from this process are no longer Dick and Jane. Each one of them is half Dick and half Jane, in its genes, in its cytoplasm, and in its mitochondria.
THIS is conjugation. It only takes place between protozoa of the same species.
Conjugation is sex without reproduction. We started with two cells and ended with two cells. They pooled their genes, but didn’t produce “offspring”. Both Dick/Jane and Jane/Dick will someday undergo mitosis and copy themselves, but Dick and Jane have ceased to be, merged instead into an amalgam.
This has nothing
to do with
propagating
The species
is continued
as so many are
(among the smaller creatures)
by fission
(and this species
is very small
next in order to
the amoeba, the beginning one)
The paramecium
achieves, then,
immortality
by dividing
But when
the paramecium
desires renewal
strength another joy
this is what
the paramecium does:
The paramecium
lies down beside
another paramecium
Slowly inexplicably
the exchange
takes place
in which
some bits
of the nucleus of each
are exchanged
Individual Selection and Group Selection, Short-term Advantage and Long-term Welfare
Why do cells do this? Let’s talk about fitness. In the short term, the race is to the swift. Reproduction is everything, especially among microbes which are always in a tight race with billions of others, and the one that reproduces fastest is the victor in Darwin’s lottery. So natural selection at the individual level motivates Dick and Jane to get on with the business of copying themselves as fast aspossible.
Why did they take time out to merge their genes? Dick and Jane individually must have thought they had a good thing going, each having survived a long while, and beaten out the competition. They each had a combination of genes that work well together. Why would they take a flier on the off-chance that their genes might do even better in some other combination? “Survival of the fittest” at its crudest level simply means that those who reproduce fastest crowd out everyone else. Sharing genes takes time and energy. You can’t afford it.
To make this less abstract: Imagine a puddle with cells swimming in it, all the same species. Suppose some of the cells—the Joneses—go straight to work reproducing, doubling their numbers, while others—the Smiths—stop along the way to have sex with other Smiths. They’re all increasing exponentially, but the Joneses grow at a faster rate. More doublings of the Joneses leads to a powerful numerical advantage. Pretty soon, the Joneses have overwhelmed the Smiths and crowded them out. The Smiths are a thing of the past, driven to extinction. We say, “the Joneses have evolved to fixation.”
Short-term individual selection says “Don’t do it! Don’t have sex!” But in the long run, the communal legacy is more robust if they DO share genes. Having many diverse combinations of genes is insurance against changes in the environment, and a high-risk investment that just might yield big dividends if the right opportunity opens up in the future. But there’s a danger that the Joneses will crowd out the Smiths in short order, and they won’t live to see the day when their robust diversity shows to their advantage.
After many, many cycles of losing sex in the short term and missing diversity in the long term, evolution stumbled on an expedient. A counter was built into the chromosomes, counting replications. Everyone is allowed to clone about a hundred times, without sharing genes. After that, without conjugation, the cell slows down and dies, stopped dead in its tracks. Every so often, every cell lineage must take time out for conjugation, or the lineage dies.
The counter is the telomere. To enforce conjugation, nature arranged for telomerase to be locked away (in paramecia and other protists) during mitosis. Each act of reproduction makes the telomeres a little shorter. Only during conjugation is telomerase unlocked, and the counter is reset, so the lineage can continue to clone.
Twenty years ago, William Clark wrote two books on this subject at a level accessible to readers of this column. Sex and the Origins of Death, followed by A Means to an End. I read both as they came out, and they had a profound effect on my thinking about evolution and aging.
Cell senescence is programmed death. Can this be an evolutionary advantage? Can programmed death evolve to protect the community from the fast crowd that doesn’t want to share their genes? Sure, there is a long-term advantage, but how was evolution so clever as to arrange this? How did it happen that telomerase came to be sequestered, available only during conjugation? I’ve looked through the evolutionary literature, and found no explanations, so I have asked this question myself, modeling with a computer simulation. The model works surprisingly well. One important feature of the model is that there is a limited reservoir of the food that cells need in order to grow. This means that the “cheaters” who avoid conjugation and reproduce faster don’t have an advantage for long, because they use up the available food store faster. Another crucial feature is that conjugation sometimes leads to combinations of genes that are more efficient at using food resources.
Here is a preliminary write-up — I plan to finish and publish this work in the near future. [Update: 2018July, finished ms as submitted is here.] Story #2: Sex and Reproduction in Plants and Animals
Half a billion years ago, there was an explosion of multicelled life. Gene sharing is not so easily arranged when there are billions or trillions of cells in each fully-grown organism. Sure, all life passes through an embryo stage, starting with a single cell. But embryos are hardly in a position to seek out a partner and share genes. So evolution needed to invent anew both the mechanics of gene sharing and a means to enforce it on individuals whose primary Darwinian motivation was to reproduce as fast as possible.
So nature took the bull by the horns (or perhaps another part of his anatomy). She laid down the law: “From now on, it takes two to tango. Anyone who wants to reproduce is going to have to share genes.”
Sex and reproduction were tied together anatomically, and the connection was so tight that no would-be cheater could get around the barriers. For some (dioecious) species, there were two separate sexes so that no single individual had the tools to reproduce by itself. In other (hermaphroditic) species, each individual could make both eggs and sperm, and there had to be barriers to self-fertilization, custom-designed for each anatomy.
Exactly how this came about is unknown. Meiosis is an operation of baroque complexity, though clearly an outgrowth of both protist conjugation and mitosis. Graham Bell (quoting Emerson) called it the Masterpiece of Nature, but neither he nor anyone proposed an evolutionary pathway that might have created it.
We know that this whole business of separate sexes and all the cellular and metabolic complexity that it entails managed to evolve, and we know that it offers no conventional advantage in terms that neo-Darwinist theory can understand. No one doubts that the link between sex and reproduction femerged from a process of evolution, but the standard mechanisms recognized by conservative evolutionary theory are at a loss to explain it.
How do we understand evolution of sex? What is the accepted explanation?
Classical evolutionary theory (neo-Darwinism) is in a bind. The theory inherited from R. A. Fisher in the early part of the 20th Century insists that there is only one mechanism of evolution, and that is one-mutation-at-a-time. Each incremental change has to provide a benefit that is capable of gradually spreading through the gene pool. In other words, all by itself and immediately it has to offer the bearers (on average) a faster rate of reproduction. On the other hand, there are numerous examples of complex adaptations (like sexual reproduction) that provide no immediate benefit for reproduction, and that require many changes to many genes in order to be functional at all. Classical evolutionary theory just says, “that’s a tough problem that we haven’t solved yet.”
But it’s more than that. The very limited repertoire of mechanisms recognized by classical evolutionary theory quite obviously can never explain the provenance of sex, or of aging, or of countless other common traits. Classical evolutionary theory is going to have to adapt or die.
I haven’t tried to model the evolution of sex because I can’t think how to do it. The problem is just too hard—all the advantage is with the cheaters, who can reproduce twice as fast because they don’t have two different sexes to support. Nevertheless, look around you—somehow nature managed to arrange most plants and animals in two sexes.
Story #3: Antagonistic Pleiotropy — a Revisionist Theory
Like sex, aging is a trait that benefits the community in the long run, but is costly to the individual in the short run. It’s not as extreme as sex—the benefit is not so essential, and the cost is much less than the cost of sex. (Two sexes cuts fitness by half, by the classical definition of “fitness”. Time and energy required for the mechanics of sex only add to the cost.) So the problem is not as severe as Story #2, but once again, nature has a problem: How to make death obligatory, so that there is population turnover and population diversity and (more important) so the population doesn’t explode past sustainable levels, leading to population crashes and extinction.
Nature’s solution was once again to tie together aging with reproduction, but the link isn’t nearly so tight and consistent as in the case of sex. In fact, population can be kept within sustainable limits either by controlling fertility or limiting lifespan, or any combination of the two, so tying longer lifespan to lower fertility (and vice versa) helps to allow for diversity and flexible strategies, while guarding against those deadly population blooms.
The name for nature’s solution is Antagonistic Pleiotropy. Fertility and longevity are coded in the genome in such a way that inheritance of lifespan and fertility are inversely linked. Higher fertility goes with shorter lifespan. Lower fertility goes with longer lifespan. As long as the two vary together in this way, the threat of population explosion can be kept at bay.
You might be thinking: pleiotropy is everywhere. We don’t need an explanation for pleiotropy, because it’s built into the way genomes are organized. Very few genes have just one mission. A web of regulation affects everything at once, so that distinct traits emerges from many genes, and every gene contributes to many traits. This is true of the way that adaptive traits are realized in nature. To think this way, you have to think of aging as an adaptive trait that nature actively wants to protect.
The Classical view of Antagonistic Pleiotropy
Contrast this with the orthodox theory of Antagonistic Pleiotropy, which has become the best-accepted theory for the evolution of aging. In the orthodox theory, genes for fertility and other traits that are highly beneficial to the individual are tightly linked to deterioration that we call “aging”. Out of the box, the genes work this way, and the forces of evolution have been unable, over half a billion years, to tease the two apart. There is a mighty motivation (says classical theory) to separate aging from fertility so that the individual can have the best of both worlds, but there are physical limitations or logical connections that make this impossible. Hence, natural selection has had to swallow the bitter pill of aging in order to get the sweet nectar of faster reproduction.
In my version, antagonistic pleiotropy is an evolved linkage, after the fact. In the standard version, antagonistic pleiotropy is an inescapable precondition, a given fact about the way genes work that evolution, with all her wiles, has been unable to evade.
How do we know that my interpretation of AP is the right one and all the theorists have it wrong?
Because the classic theory requires that every “aging gene” must have a benefit that more than compensates, and after 30 years of genetic experiments, pleiotropic costs.have been identified for only about half of the known aging genes.
We’ve seen that evolution is capable of some amazing feats. It just doesn’t pass muster that evolution has been trying to find a pleiotropy bypass for half a billion years but doesn’t seem to be able to find one.
Because some of the best-known cases involve quasi-pleiotropic linkages that can be broken in the lab. It’s just not that hard to have your cake and eat it, too. The first example was AGE-1, the first bona fide aging gene to be discovered (in lab worms, 1989).
When you look at the actual mechanisms of pleiotropy, many of them don’t seem to be functionally essential, but involve unexpected connections between unrelated functions. The most recent example is that methylation aging seems to be inversely related to telomerase expression.
Of these 3 stories, the story of evolved Antagonistic Pleiotropy (#3) is the easiest to model and simulate, which is to say that the model requires few assumptions and works to evolve pleiotropy without a lot of adjustment or tinkering. This alone gives me confidence that AP is evolved, and that the usual interpretation for the meaning of AP is upside down.
I have been working to turn my computer model into an academic article, and a draft of the paper, not yet submitted, is posted here. [Update: 2018July, finished ms as submitted is here.]
Part II: Why We Should Trust the Methylation Clocks to Measure Aging
Last week, I proposed that methylation age could be used to measure the benefits of putative anti-aging interventions. This procedure has the potential to slash the cost and the duration of testing. The reason is that we don’t have to wait for a small percentage of experimental subjects to become sick or die. The vast majority of subjects in a human anti-aging trial give us no information whatever. In contrast, with the aging clock, every experimental subject is a data point, and the effect on his aging might be measured in a year or two.
The proposal depends critically on the assumption that whatever slows aging will slow the methylation clock, and the converse: whatever slows the methylation clock slows aging. Some people will find this hard to believe, because their fundamental conception of aging is an accumulation of damage, so that any association with methylation will be incidental or worse! (What if the changes in methylation that accompany aging tell the story of the body’s increasingly powerful efforts to repair the damage from aging?)
But for those of us who come to the table open to the idea that aging is an epigenetic program, a close (causal) association between methylation and aging seems utterly expected. For decades, developmental biologists have assumed that development in childhood is driven by age-dependent gene expression. The only thing that prevents us from seeing that the same is true about aging is a kind of prejudice from evolutionary theory that I have described in my book and in this blog.
4. Brief History of the Horvath Clock
Of many biochemical markers that cells use for epigenetic control, methylation of CpG sites is best studied. If you know what that is all the better; if you don’t, all you need to know is methylation is a modification of the DNA by adding a CH3 group to Cytosine residues in a promotor region adjacent to a gene. Regions with heavy methylation tend to suppress expression of the (usually) adjacent gene. Methylation isn’t the only means by which gene expression is controlled — there are many others. But it is far the best-studied and, given present technology, it is the only epigenetic marker that can be routinely measured, for a few hundred dollars in a small sample of blood, urine, or nanogram-scale biopsy of other tissue.
The clock was developed by Steve Horvath at UCLA, and first published in 2013, built on an idea of Teschendorff from a few years earlier.. He identified patient records for methylation measurements of tissue samples from 8,000 individuals, with associated ages. Methylation is recorded as a number between 0 and 1 for each Cytosine, indicating the proportion of that site that is methylated. He scanned the entire genome for sites that changed most with age, and varied least from one tissue type to another. In this way, he identified 353 sites, and optimized a set of 353 multipliers, such that multiplying levels of methylation at each site by each multiplier and adding the products produced a number that could be mapped onto chronological age. About 45% of the multipliers are negative (sites losing methylation with age) and 55% positive (gaining methylation).
The original Horvath clock correlates 0.95 with chronological age. The standard error in predicting any one individual’s age is + 4 years. Averages of N individuals increase the accuracy of the clock by √N, so that the average of 100 individuals is accurate to 0.4 years. (This is a general statistical principle that is useful to remember.) For our purposes, the relevant question is: measuring the same individual at two different times, how accurate is the difference in Horvath age compared to the elapsed time? There is no data on this yet, but we might safely assume that it is well under 4 years, since standard error of 4 years represents mostly individual departures from the average.
Five years after Horvath’s original publication, there are several other clocks based on methylation. Just this spring, Horvath has developed a new clock, not yet published, which, to my knowledge, is the best standard we have. This is the Levine/Horvath clock. It is based on 513 methylation sites and it is calibrated not to chronological age, but to a tighter measure of age-based health, derived from blood lipid profies, inflammatory markers, insulin resistance, etc, which Horvath calls “phenotypic age”. Consequently, it is less well correlated with chronological age than the original, but it is better able to predict mortality than either the classic Horvath clock or chronological age itself. By this measure, the scatter has been greatly reduced.
There is statistical evidence that the Levine clock reliably reports phenotypic age, and there is theoretical reason to believe that what the clock measures is close to the root cause of aging.
5. Statistical evidence that the Levine Clock=PhenoAge reliably measures biological age
What I find most convincing is the meta-analysis based on historic data. Levine and Horvath use old, frozen blood samples to calculate a Horvath and Levine Ages as it was at some past date. These are people who have died since the blood was drawn, and Horvath Age accurately “predicts” the remaining life expectancy of the subjects. [Chen, Aging 2016]. There is less data available for the new Levine clock, but strong indications it performs much better than the Horvath clock for this purpose.
In addition, many of the life styles that promote long life have been confirmed to slow the Levine clock, while, conversely, obesity and high blood pressure and insulin resistance have been found to accelerate aging as measured by the Levine clock.
Epigenetic age correlates with progression of Alzheimer’s and Parkinson’s Disease [Levine 2016]
Menopause moves the methylation clock forward. Early menopause is associated with accelerated methylation aging, and late menopause with younger methylation age.
Epigenetic age is accelerated by obesity, blood sugar, insulin, and inflammation
Epigenetic age is retarded by carotenoids*, exercise, education (!), and by diets high in vegetables, fruits and nuts.
Stem cell transplants lower epigenetic age more dramatically than anything (from a study of leukemia patients [Stolzel 2017]). Epigenetic age is set back ~8 years for a short period, but then accelerates to a set-forward a few years after treatment.
6. Theoretical foundation of the Horvath Clock
The original Horvath clock was developed by a statistical process that took into account only chronological age. But Horvath age turns out to be a better predictor than chronological age for risk of all the diseases of old age. This is powerful evidence that methylation is measuring something fundamental about the aging process. If an individual’s methylation age is higher or lower than his chronologial age, the difference is a powerful predictor of his disease risk and how long he will live. This can only be true if methylation is associated with a fundamental cause of age-related decline.
An emerging theory the last 7 years is that aging procedes under epigenetic control. De Magalhaes, Rando, Blagosklonny, Johnson and Mitteldorf—all have independently proposed an epigenetic basis for aging. The root cause of aging—the reason our bodies are different at age 70 compared to age 20—is that different sets of genes are expressed at different times of life. This priniciple is already well-accepted for growth and development [ref, ref]. During formation of the body in utero, gene expression rapidly changes, and in early childhood, the growth and mathuration of the body are widely agreed to occur under epigenetic control. But now we know that much of the change in methylation is continuous, from development through aging [ref]. I call this programmed aging. Blagosklonny hedges and says “quasi-programmed”. The difference is about evolutionary purpose and whether function is related to natural selection. My view is that we are programmed for a fixed lifespan for the stability of the community. Blagosklonny’s is that the epigenetic changes that start in development continue afterward through a kind of inertia because there isn’t enough natural selection to turn these changes around.
But for the sake of the reliability of the methylation clock in evaluation of anti-aging interventions, these two perspectives converge: they both support the expectation that methylation age will be an excellent criterion for trying and judging new ideas and combinations of old ideas.
Parabiosis experiments support the idea that factors circulating in the blood have a deep effect on the age of the body. This is indirect support for the epigenetic foundations of aging, because these blood factors come from gene expression in cells–especially but not exclusively endocrine cells.
7. Counter-arguments
A) ‘Epigenetic drift’ —Many authors still write about changes in methylation during aging as “epigenetic drift”. For those who cannot accept the idea that aging is programmed, it is much more palatable to imagine a loss of order in gene expression, a randomization of gene expression. Indeed, this is true. It is part of the story that gene expression doesbecome more random with age. But it is also true that there are specific gene expression changes associated with aging–the methylation clock is based on such programmed changes.
B) Perhaps gene expression changes are a response to damage, the body’s attempt to mitigate aging. This is the suspicion that haunts the aging clock. If this is the case, interventions that thwart the mitigation would come out looking like age reversal, but in fact they’d have the opposite effect, increasing risk of disease and mortality. Support for this idea comes from the prejudice that says “the body would never purpposefully destroy itself.” But there is no evidence for this idea, and in fact many of the programmed changes have been shown to be detrimental. For example, signals for inflammation are increased, DNA repair is slowed down, and the anti-oxidant metabolism is suppressed.
“DNA PhenoAge acceleration was found to be associated with increased activation of pro-inflammatory and interferon pathways and decreased activation of the transcriptional and translational machineries, the DNA damage response and nuclear mitochondrial signatures” [quote from Horvath 2018; footnote is to Levine 2018
C) Not all anti-aging interventions affect the Levine or Horvath clocks. This is a substantial problem if it turns out that there are real anti-aging strategies that work, and yet the Levine clock won’t tell us that they work. But we don’t really know this yet, because we don’t really know what works. “For example, within a 9-month follow-up period, the substantial weight loss resulting from bariatric surgery was not associated with a reduction in epigenetic age of human liver tissue samples” [quote from Horvath 2018; footnote is to Horvath 2014] To the extent we think that bariatric surgery is a legitimate anti-aging strategy, this is a problem
8. Improvements and adaptations of the Horvath clock
The “clocks” we’re talking about are really mathematical operations. Given the output of a blood (or urine) test that reports what percentage of the DNA is methylated at each of hundreds of thousands of different CpG sites, the “clock” is a computer program that distills this information down to a single number, the predicted age.
The Levine clock is a substantial improvement on the original Horvath clock, attained by calibrating it against health indicators and not just chronological age. For prediction, it leaves its predecessors in the dust.
There are three more ways in which the methylation age test can be improved, and I have begun working with the Horvath lab to do the number crunching in support of these changes.
A) The original clock and all its successors have thus far been based on combining information from 353 different methylation sites in the simplest possible way. They simply have 353 different multipliers. It is these 353 (positive and negative) numbers that have been optimized by the statisticians, so that each multiplier can be multiplied by each methylation, and the 353 products are added up to make a single number that indicates age. My suggestion is to combine the 353 sites in a more flexible way. Some change rapidly during youth and then remain constant. Some change continually over a lifetime. Some don’t change much at all until aging sets in. There is no reason that all the 353 sites have to be treated the same way. Using non-linear math that’s just a little more complicated, the 353 sites can be tracked in a way that corresponds to their peculiar lifetime trajectories. This will improve the clock’s accuracy for any application.
B) The clock might be specialized to the application of testing anti-aging effects on individual humans, i.e., comparing biological age for the same individual at two different times. Some of the scatter in the plot of DNAmAge is due to variation from one individual to another, and some is due to other random factors that don’t depend on the individual. In the past, there was little data available for the same individual at two different times, but this is changing, and now it is feasible to separate the two kinds of scatter. The clock can then be specialized to report age differences even more accurately.
C) Again, for the particular application proposed, there is no need for a clock that works generally on any age, from pre-birth to centennarian. If all of the people in the study are between the ages of 50 and 70, then the clock might be specialized to be more accurate in this age range, at the expense of losing accuracy for younger and older subjects–who aren’t part of the study. It may be worthwhile to take this idea even further and have four sub-specialized clocks, calibrated for ages 50-55, 55-60, 60-65 and 65-70.
In my brief experimentation with the data, I was able to raise the correlation from 95% to 96% using technique #1. I’m guessing that with further work it can be raised to 98%. The reason that it pays to do this is that the cost of a human study depends on (A) how many people are studied and (B) how long a time they are followed. As the scatter in the data is reduced by better statistical techniques, we can find out what we need to know with fewer subjects and a shorter study time. Raising the correlation from 96% to 98% will reduce the number of subjects needed for the experiment by a factor of 4. Alternatively, for the same effort and expense, we wil be able to derive more information.
If we can indeed construct a clock with 98% accuracy, a new benefit will be available: It will be accurate enough to distinguish changes for a single individual with no statistical averaging necessary. This will be a gateway to individualized medicine. There will always be treatments that work for some people but not others, and the future of medicine is connected to knowing what works for you as an individual. Each of us will be able to use the methylation clock to know how we are doing. You can try a new supplement for a year and if it doesn’t work for you as an individual, you’ll know it and switch to trying something else next year.
_____________
*carotenoids are molecules related to vitamin A, but vitamin A itself does not slow the aging clock.

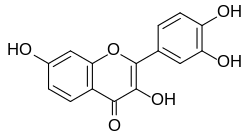
 (MEF stands for Mouse Embryonic Fybroblast, the cells that were cultured in the screening experiment.)
(MEF stands for Mouse Embryonic Fybroblast, the cells that were cultured in the screening experiment.)








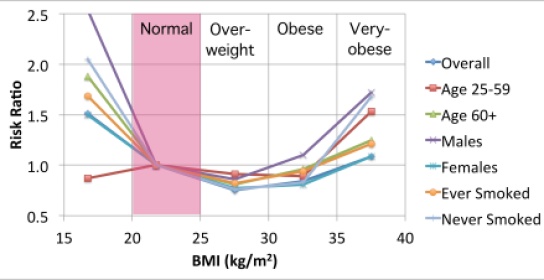






 4. Brief History of the Horvath Clock
4. Brief History of the Horvath Clock