
Senolytic drugs have been the most promising near-term anti-aging therapy since the ground-breaking paper by van Deursen of Mayo Clinic published in 2011. The body accumulates senescent cells as we age, damaged cells that send out signal molecules that in turn modify our biochemistry in a toxic, pro-inflammatory direction. Though the number of such cells is small, the damage they do is great. Van Deursen showed that just getting rid of these cells could increase lifespan of mice by ~25%. But he did it with a trick, using genetically engineered mice in which the senescent cells had a built-in self-destruct switch.
After that, the race was on to find chemical agents that would do the same thing without the genetically engineered self-destruct. They must selectively kill senescent cells, while leaving all other cells unharmed. It’s a tall order, because even a little residual toxicity to normal cells can be quite damaging. Before last week, the two best candidates were FOXO4-DRI and a combination of quercetin with dasatinib.
I’ve written in the past (here and here) that senolytic drugs are our best prospect for a near-term lift on the road to anti-aging medicine.
Last week, a large research group affiliated with the original May Clinic team published findings about fisetin, the latest and greatest candidate for a senolitic pill, another flavenoid, very close in structure to quercetin. 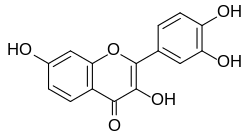
They grew senescent and normal cells in a test tube, then tested 11 different plant-derived chemicals for power to kill the one while leaving the other unharmed. The winner was fisetin.
 (MEF stands for Mouse Embryonic Fybroblast, the cells that were cultured in the screening experiment.)
(MEF stands for Mouse Embryonic Fybroblast, the cells that were cultured in the screening experiment.)
Fisetin is especially interesting because it is cheap, easily available, widely-regarded as safe, but not nearly as well studied as quercetin.
They took the winner, fisetin, and subjected it to a series of tests. They began with in vitro (cell culture) tests and proceeded to in vivo tests with live animals, culminating with an impressive life span assay in mice.
(The runner-up was curcumin, less interesting perhaps only because it has already been extensively studied. The curcumin molecule is unrelated to quercetin or fisetin, and is not a flavenoid. I can’t help but wonder if they had subjected curcumin to the same thorough testing that they reserved for fisetin, how would curcumin have fared?)

curcumin
The paper’s principal findings were:
- Fisetin has lower liver toxicity (at equivalent doses for senolytic benefit) than any of the other senolytics tested so far.
- Fisetin reduces pro-inflammatory signaling in a short course given to mice and in long-term experiments where fisetin was added to the mouse chow.
- Fisetin reduces number of senescent fat cells in a short course given to mice.
- Mice fed fisetin for long periods had much more glutathione than control mice. (Glutathione is one of very few marker molecules that seems to be wholly beneficial.)
- Most impressively, mice fed fisetin late in life lived 10-15% longer than control mice. This represents a 50% increase in the remaining lifespan after the intervention.

What we know and what we’d really like to know
We’d like to know, do humans who take large doses of fisetin live longer? Do they have toxic side-effects? These questions require decades to answer.
Does fisetin reduce age markers in humans, especially methylation age? This is a feasible study, since the test is mature and safety of fisetin is fairly well established for short courses. Perhaps this experiment is being considered; I’ve written to the corresponding authors of the most recent study, in case they haven’t already thought of it. This test would not be definitive because we know that methylation age is not perfectly correlated with biological age; but if positive it would confirm both that fisetin is accomplishing epigenetic rejuvenation and that methylation tests were correctly informing us of this; a negative result would be ambiguous.
Episodic Dosing
It makes sense that senolytics should be taken periodically, not continuously. A high dose can be toxic to existing senescent cells, and then getting out of the way, it can allow normal cells to recover from any damage. This sounds like good theory, but different dosing regimens have not been tested experimentally. In fact, the new paper reports positive results from both high episodic dosing and lower everyday dosing.
The Mayo group had previously tested fisetin, and found it effective in killing some kinds of human senescent cells but not others. In previous tests, fisetin was found to be effective in senescent fat cells (pre-adipocyte, white adipose tissue), and that is where it was primarily tested in the new studies.
Authors’ comments
They note that the episodic treatment and short half-life suggest that the benefits of fisetin come from its senolytic action, rather than other actions as an antioxidant and signal molecule. They emphasize that clearing senescent white blood cells and making room for new, active white blood cells are activities that enhance the benefits of fisetin, since white blood cells contribute to clearing the remaining senescent cells.
Fisetin has previously been shown to have anti-cancer activity and to inhibit inflammatory signals directly. Here is a review of benefits of fisetin from three years ago. Drugage lists just two previous lifespan studies with fisetin, with encouraging results from yeast and fruitflies.
The Bottom Line
If we choose to take fisetin at this stage in the science, we are early adopters, and our main concern ought to be safety. There is little doubt that killing senescent cells will be beneficial. But what is the toxic burden of fisetin, and what dosage can we safely take without risk of damage to normal cells? The current study covers a lot of ground but doesn’t answer this question, apparently because they are convinced that fisetin is quite safe.
Strawberries, apples, grapes, and onions all contain fisetin, but at low levels compared to a senolytic dose. For example, the highest food concentration, 160 ppm, is found in strawberries. A half pound of strawberries yields 36 mg of fisetin. We’re still guessing at the therapeutic dose, based on mouse studies, and the experimental dosage in human trials is about 1,000 to 1,500 mg (based on this clinical trial), the content to 30-40 pounds of strawberries on each of two consecutive days.
In the best cases, fisetin was shown to reduce senescent cell burden by 50% and up to 75% in cell cultures. This is a good start, and encourages us to think we can do better by combining fisetin with other agents, or perhaps with fasting.
Also reported today,
Clearing Senescent Cells From The Brain In Mice Preserves Cognition
It sounds impressive, but I’m not impressed. First, mouse models of Alzheimer’s have been discredited repeatedly. Mice don’t naturally get AD, so they have to be genetically engineered to do so, and the genetically modified mice don’t share the deep causes of human AD. Time and again, treatments have been found effective in the mouse model that fail to translate to humans. Second, the treatment used in the study to kill senescent brain cells also relied on another genetic modification, and would not be applicable to humans.
My guess is that effective senolytic agents for humans will be available within a few years, and that they will decrease risk of all age-related disease, including Alzheimer’s. But this study does little to advance us toward that goal.


