
“Cancer is a genetic disease. Its primary cause is mutagens in the environment, abetted by time and bad luck. A cell is controlled by the chromosomes in its nucleus, and when just the wrong combination of mutations happens to occur, a cell can begin to grow and multiply uncontrollably. The next crucial step occurs when the cell acquires the ability to travel through the bloodstream and implant somewhere else. The whole pathway from errant cell to malignant cell proceeds via chance mutations. From inception to metastasis, cancer is driven by genetics.”
This theory of cancer is more than 100 years old, but it didn’t become the dominant view until the 1950s, when, after Watson and Crick, genes assumed an exalted position in the study of biology. The “somatic mutation theory” continues to dictate the course of cancer research and treatment today.
It is uncontested that cancer cells have abnormal chromosomes. Dozens of different mutations have been found in malignant cells. They have been catalogued as different oncogenes, and because they are so different in their functions, cancer has been re-conceived from a single disease to a category containing many different diseases with similar symptoms.
Are mutated genes the root cause of cancer? Toxins that commonly break DNA (teratogens) are also found to cause cancer (carcinogens). Radiation, ditto. “Ionizing” radiation packs enough wallop in each photon to break a chemical bond, and is associated with cancer, while non-ionizing radiation (visible, infrared, and radio waves) is not mutagenic and generally not carcinogenic*. This has been taken as powerful circumstantial evidence for the prevailing theory.
A direct answer to the question of whether cancer originates in the nuclear DNA is available from an experiment that is simple in principle: Swap nuclei between two cells, one normal and one malignant. Take the mutated DNA out of a cancer cell and put it in a normal cell, to see if it becomes malignant. Take the un-mutated DNA out of a normal cell and put it in a cancer cell to see if the cell is rescued and restored to health.
This experiment has been technically feasible for more than 30 years, and indeed Barbara Israel and Warren Schaeffer actually performed both experiments at UVM and wrote them up in 1987 [ref, ref]. The results were exactly the opposite of what was expected: The cell with normal cytoplasm and cancerous nucleus was normal; the cell with normal nucleus and cancerous cytoplasm was cancerous. This result has been confirmed in other labs [reviewed by Seyfried, 2015]. Still, the genetic paradigm has a stubborn grip on cancer research and treatment to this day.
An alternative theory of cancer as a metabolic disease was put forth by the Nobel polymath Otto Warburg in the 1930s. The principal proponent of this theory today is Thomas Seyfried of Boston College. Seyfried cites evidence that damage to the nuclear DNA, conventionally thought to be a root cause of cancer, is actually an effect of the damaged mitochondria and irregular metabolism. “The metabolic waste products of fermentation can destabilize the morphogenetic field of the tumor microenvironment thus contributing to inflammation, angiogenesis and progression.”
Respiration and Fermentation
Every cell in our bodies (and almost every cell in all eukaryotes everywhere) makes uses of energy in the form of ATP, adenosine triphosphate. ATP is manufactured in the mitochondria, usually by a controlled burning of sugar to form CO2 and H2O. Highly energy-intensive cells such as muscles and nerves have thousands of mitochondria in each cell. The word “respiration” in this context is used to mean burning sugar in an efficient energy conversion process, yielding 38 ATPs for every sugar molecule. But when oxygen is scarce, perhaps because you’re breathing as fast as you can or sprinting in deep anaerobic mode, another process can be used to rapidly convert available sugar stock to lactic acid, requiring no oxygen at all, but yielding only 2 ATPs per sugar molecule. The latter process is called “fermentation”. (This observation explains the extraordinary effectiveness of interval training (sprints) for weight loss.)
Warburg was among the first to notice [1931] that most cancer cells use fermentation rather than respiration as an energy source. Metabolic studies pointed to damaged mitochondria in tumor cells that had become inefficient in producing sufficient energy through respiration. He theorized that impaired mitochondrial function is the root cause of cancer. In fact, Warburg did some of the early work establishing the role of mitochondria as cellular energy factories.
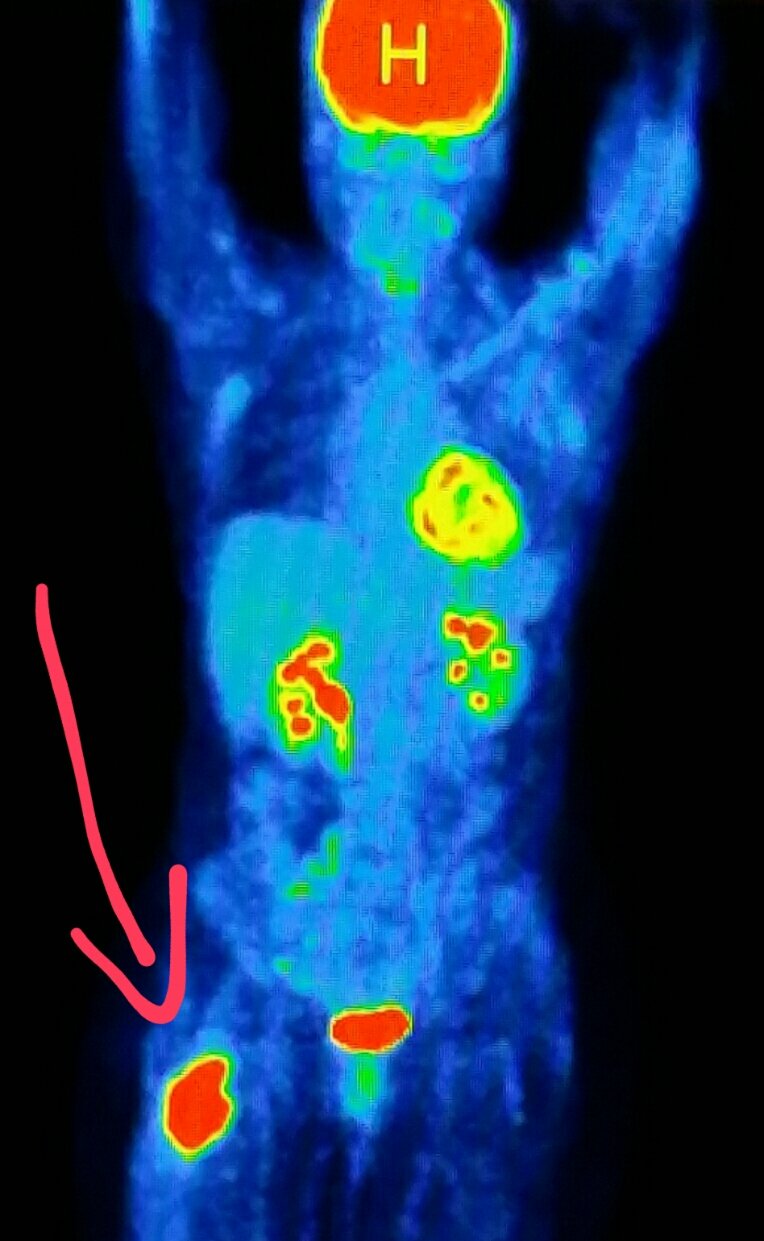
So most cancer cells are sugar addicts. They consume enormous amounts of sugar, both because they are actively growing and dividing, and also because they use sugar so much less efficiently than normal cells. A PET scan can be used to visualize concentrations of sugar in the body, and PET technology is often used to locate tumors.

Sugar is easily made from carbohydrate foods, and when you eat a diet containing carbs, sugar is the fuel of choice. Ketones are an alternative fuel used by the body when burning fat, either stored fat or ingested animal fat or vegetable oils. (Medium chain saturated fatty acids like coconut oil seem to be most effective in inducing metabolic ketosis.) Unlike sugar, ketone bodies cannot be fermented. They generate ATP energy only through oxidative respiration in the mitochondria.
The logical question:
Are zero-carb diets an effective treatment for cancer?
Some well-known cancer drugs (Gleevec, Herceptin) already target the fermentation metabolism. Acarbose has been proposed but not yet tried. But might it be safer and more effective to starve cancer cells by cutting carbohydrates in the diet to zero? There is a robust literature suggesting, “yes” [e.g., ref, ref, ref, ref, ref, ref, ref] but so far the results have been less than earth-shaking.
A search of ClinicalTrials.gov yields 25 trials of ketogenic diet variants for cancer treatment. Most are in early stages, 5 have been completed, 2 have results. In this study, the ketogenic diet, with or without chemotherapy, did not cure glioma. This small study found modest benefits in a variety of advanced cancers. These results are consistent with many mouse studies, in which some benefit was recorded from the ketogenic diet, but not a dramatic difference. The most encouraging results I have found was a study in which 9 of 11 mice treated with a combination of radiation and a ketogenic diet were cured of brain cancer. Clearly, this is no miracle cure, but it’s too early to give up–we’re just figuring out how to make the diet work, and it has not yet been tried except at late stages, after all else has failed.
Fasting shows more promise than ketogenic diets. (Perhaps fasting lowers blood sugar even more than ketogenic diets.) A series of studies by Valter Longo make the case that fasting simultaneously sensitizes cancer cells to chemo or radiation and de-sensitizes normal cells.
Seyfried has proposed a “press-pulse” system based on this vulnerability, targeting the glucose metabolism and the glutamine metabolism with hyperbaric oxygen. Besides glucose, glutamine is also a major fuel for tumor cells. Drugs will be required to target glutamine, as glutamine is the most abundant amino acid in the body and can be easily synthesized from glutamate. Hyperbaric oxygen requires a patient to be enclosed in a pressurized oxygen chamber or room filled with pure oxygen at 2.5 x atmospheric pressure. There is one highly encouraging case report for the success of this triple combination—hyperbaric oxygen, glucose inhibitors, and low-dose chemo—in which a late-stage, resistant breast cancer is driven to total remission.
Last week, a research paper from Duke U suggested a target for attacking the fermentation metabolism of cancer cells, and a marker for identifying which cancers are likely to be sensitive to it. The research group of Jason Locasale found a protein called GAPDH which switches to the fermentation metabolism, and a compounded called koninjic acid, extracted from fungi, that inhibits GAPDH. They have tested koninjic acid extensively in cell lines, and have begun testing in live mice. Whether such drugs are more effective than simply restricting glucose is a topic for investigation.

Mito-targeted Cancer Prevention
Supplements that promote mitochondrial health include CoQ10, PQQ, mitoQ/SkQ, alpha lipoic acid (ALA), carnitine, and melatonin. Can they lower risk of cancer? So far, we have just a few hints; this is a promising area for research.
CoQ10 was studied in the 1990s as a cancer treatment, with some encouraging results [ref]. PQQ has been shown to kill cancer in vitro [ref]. One mouse experiment looked at ALA as part of a cancer treatment [ref]. Use of carnitine remains theoretical [ref]. Most has been written about melatonin [ref, ref, ref], but even here, there is no epidemiological evidence.
The Bottom Line
All the evidence for radiation and other mutagens causing cancer might be re-interpreted in terms of mutations to mitochondrial DNA. (Mitochondria live in the cytoplasm, outside the cell nucleus, but they have a bit of their own DNA and ribosomes for transcribing it.) Damaged mitochondria can also cause cancer even when their DNA is intact, and Seyfried (after Warburg) makes a strong case that mitochondrial damage is the root cause of cancer. Inflammation is probably the single worst source of mitochondrial damage. Do we need one more reason to minimize inflammation? Viruses often target mitochondria for their own ends, and this may explain cases in which viral infections are associated with etiology of cancer.
The insight that mitochondrial damage is the root cause of cancer (preceding nuclear mutations) also has broad implications for cancer prevention. As for treatment, there have been a few disappointments and also some promising pilot studies, especially in combining glucose deprivation with radiation or chemo to finish the job (“press-pulse”). This is a research field that deserves much more attention.
__________
*There are exceptions to both these generalizations. There is controversy whether ionizing radiation at low dosages causes cancer [ref]; and cell phones (non-ionizing) have been linked convincingly to cancer risk, presumably by a different mechanism than breaking chromosomes [my column last year].
I sent a draft of this column to Thomas Seyfried, who was kind enough to edit it in detail and add references of which I was unware.
I was led to this subject by my co-author’s publisher, Chelsea Green, publishers of
Tripping over the Truth, by Travis Christofferson.
Discover more from Josh Mitteldorf
Subscribe to get the latest posts sent to your email.
Has anyone looked at SKQ1 or MitoQ wrt cancer? What about nicotinamide riboside?
There is supposed to be an NR Phase 1 cancer trial somewhere, but now it’s not turning up for me. Anyone else see that?
The NR cancer work is at Scripps. Don’t know how far along.
I referenced a paper below where they used MITOTEMPO and mitoQ to stop cancer metastasis. Didn’t actually destroy the original cancer, just stopped it spreading.
HI there Josh>.I have always thought that cancer was a result of a failure of apoptosis to occur correctly. Why? During mitosis and apoptosis in both cases the genome is demethylated so DNA can be copied duirng mitosis , or snipped to pieces during apoptosis. You see methyl groups(5mC) attached to DNA at restriction sites prevent restriction enzymes from cutting the DNA. So my guess has always been that evolution probably just conserved the beginning stages of mitosis and later evolved an alternative pathway in the mitosis cycle which leads to apoptosis. So my thinking was that by using this machinery, if for some reason the DNA fragmentation program does not kick in to cause apoptosis, the whole process gets stuck in the “on” mitosis mode, and the cell just keeps replicating instead of self destructing.Now checking on how mitochondria are involved in apoptosis and mitosis, you see they play a key role in controlling both processes…For example>> Mitochondrial fission in apoptosis
Richard J. Youle & Mariusz Karbowski
Abstract
Mitochondria fuse and divide continuously within cells to form a dynamic network. One of the steps in apoptosis is the fragmentation of mitochondria, and recent evidence indicates that the mitochondrial fission machinery actively participates in the process of programmed cell death.AND
MITOCHONDRIA FUSE
Many protein machines are required in the process of mitosis. Protein machines are powered by energy molecules called ATP (adenosine triphosphate). The mitochondria are bean-shaped organelles that specialize in producing lots of ATP. The S phase of interphase is when the cell makes a copy of its entire DNA, which will be evenly divided during mitosis. But before the cell commits to entering S phase, it must have enough ATP molecules. Before a cell enters the S phase, its mitochondria will fuse with each other to form a network. This network produces much more ATP than individual mitochondria can.
So I havent done much research on this idea>>just thought I would throw it out for y’all to chew on.
Just goes to show how important mitophagy is. If fusion gains too much prominence over fission, and we know in a well fed state it does, then mitophagy is impaired (mitochondria fused so less functional ones can’t be detected through low intermembrane potential, plus are too big for autophagsomes), and if what you are saying is right, apoptosis is impaired too.
We also know calorie restriction improves mitochondria function and lowers the incidence of cancer. So maybe it is not the mounting up of random nuclear mutations that causes cancer, but the slow and steady decline in metabolic health caused by too many calories? From what we know of the very low cancer rate in Egyptian Mummies, this has some support.
One other point of interest would be to look into the health of mitochondria in senescent cells to see if the short telomeres – senescent arrest – cancer escape from arrest could be explained through a mitochondrial mechanism.
I think there is a lot to be said for mitochondria or a being the cause of cancer.
Not necessarily a contradiction. Mitochondria are the vehicles that implement apoptosis.
It is strange because we know apoptosis is triggered by a large increase in ROS. Here is a paper where they enhanced apoptosis using Trichostatin A (which incidentally is a strong telomerase activator in non cancerous cells)
‘Trichostatin A Targets the Mitochondrial Respiratory Chain, Increasing Mitochondrial Reactive Oxygen Species Production to Trigger Apoptosis in Human Breast Cancer Cells’, Plos One, March ’14
But increases in ROS also trigger metastasis, see:
‘A Mitochondrial Switch Promotes Tumor Metastasis’, Cell Reports, Aug 2014
They were able to block this with mitochondrial targeted antioxidants, which is quite exciting.
So mitochondria are clearly very important in cancer. Increases in ROS can either hinder or help cancer, depending on the stage of the disease, perhaps?
Hi Josh,
One really great column. Will give it great deal of study.
If I’m not mistaken, there are some clinical trials in progress for transfusing exogenous mitochondria into patients with diseases characterized by mtDNA depletion. You wonder if this might be a strategy in cancer therapy if cancer as a mitochondrial disease is correct.
“destabilize the morphogenetic field of the tumor”
(?) That isn’t a sentence that means anything in biology… “morphogenetic field” is the vague mystical idea that we used before discovering chromosomes and DNA…
The 1987 experiment looks really interesting, and the result is indeed opposite of predicted by standard theory (which could probably be described as “not meaning anything” either 😉
Yet if you google “morphogenetic field of the tumor” you’ll find various papers using the term. ; – )
In any case, it’s great to see the metabolic theory of cancer getting some coverage. I’ve been following (as a layman) the work of Thomas Seyfried and Dominic D’Agostino etc for a few years now.
Cheers,
Nick.
Apparently some people still use “morphogenetic field” to mean “cells in a line of descent that are meant to make a particular structure (e.g. a leg).” It’s not helping me follow what they’re saying, because the most common use is “mystical energy field that tells cells how to develop”. No complaint if they can actually explain what that means, but usually it’s gibberish like this:
https://www.sheldrake.org/research/morphic-resonance/introduction
Seyfried has sent me a paper explaining what a morphogenetic field is.
– Josh
thanks.
From a purely medical clinical viewpoint, I find that cancer is very very complicated on many levels. There is a clear genetic predisposition, telomerase activation and critically short telomeres, a blocking of tumor inhibitors ( p 53), an acidic growth environment, chronic inflammation, glycolysis for energy, angiogenesis, mutations, mitochondrial dysfunction, loss of apoptosis, cell type , connective tissue environment, IL 6 and IL8 , oncogenes, and immune system failure. That’s how I view it as a clinician, and I doubt that there is any one answer to this horrible problem.
Hi Josh,
Cancer theory you presented fits in very well with my theory of cancer presented in prior post, in particular reason for fermentation, cytoplasm control and relation to mitochondria.
First part of theory is cancer is normal growth from prior to 500,000 million years ago, prior to Cambian period. That was before plants and before oxygen rich atmosphere; life was fermentation, unlimited telomerase, no aging, cells were immortal.
Second part is cell life is organized in the cytoplasm not the nucleus. This can be seen with TOR as an example. TOR controls growth and development. The nucleus is like a file cabinet with blue prints for all proteins. However, the command and control is in the cytoplasm and through transcription and translation, TOR orders through messages sent to nucleus the proteins to be produced. (example of control in cytoplasm, not saying TOR related to cancer)
Third: going back 500,000 years, there would have been a substance in cytoplasm I will call substance J, which controlled the genes which controlled the metabolism which was cancer-like, by current standards.
Fourth: Around 500,000 million years ago, it is then postulated there is a sea change from fermentation like metabolism to oxidative phosphorylation centered around mitochondria. This would have been coupled with putting substance J in chains and locking up all the genes under control of substance J as switched from cancer-like growth to organized growth typical of normal tissues we see today.
Fifth: The repression of substance J is organized by mitochondria; but
Substance J persists under restraint in the cytoplasm and all the genes for cancer-like growth persist in the nucleus.
Sixth: Cancer results from failure of mitochondria (mutations, damage) to continue inhibition of substance J. Substance J becomes unchained. Substance J then unlocks all the genes in nucleus for cancer-like metabolism that have been conserved from 500,000 million years ago.
This explains why the substance which can promote cancer is in cytoplasm and not the nucleus and explains the link between cancer and mitochondria.
Reason substance J has not identified; because looking for it in nucleus and not in cytoplasm and substance J is normally inactive. However, I think your first references show that an active substance J which can unleash cancer must reside in cytoplasm of cancer cells.
Because cancer represents a form of life present 500,000 million years ago and all the genes for cancer are already present in nucleus; it explains why cancer can suddenly emerge as such a robust form of life. It is not something original being created de novo with each new cancer.
In summary: cancer starts in the mitochondria, which releases control of substance J in cytoplasm, which then unlocks all the blueprints for cancer preserved in nucleus.
Josh,
As usual a very well researched post with many relevant studies cited for deeper research. Cancer can be beaten just as aging can be cured – just a matter of time. For example CAR-T technology is showing phenomenonal responses in blood cancers 8 out of 10, 9 out of 10 going into remission without the horrible side effects of chemo.
Regarding interesting observation on respiration and fermentation – fasting and intermittent fasting have shown better results than cyclic or long term ketogenic diet against cancer. Longo has many interesting studies on fasting and cancer one of them is ‘Fasting and Caloric Restriction in Cancer Prevention and Treatment’ 2015.
Interesting post. Another way to look at this is to check the odds of cancer for people affected by a mitochondrial mutation. I am one of them, so I have looked at this in the past. This is[1] a good epidemiological study about this. In this case, the conclusion seems good for me:
“Conclusions: Patients with mitochondrial dysfunction do not appear to be at increased risk of cancer compared with the general population.”
[1] https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4366902/
From the Link Luis provided:
(“conclusion, the results of the present study do not support the existence of an increased risk of cancer in patients with perturbed mitochondrial function. Although the opposite scenario is not contradicted by our results, that is, that cancer-induced alterations of cellular metabolism can cause secondary mDNA aberrations that subsequently start a vicious circle of further malignant transformation (Gogvadze et al, 2008), impaired mitochondrial function per se does not appear to cause cancer.”)
Interesting Study
Hi Josh,
As regards your theory of cancer and mitochondria, I had a post yesterday explaining how this could have evolved and a theory of cancer I thought very similar to your theory. Could you indicate if we are in agreement as to following points:
1. Cancer begins in cytoplasm, not the nucleus.
2. The first step in cytoplasm where cancer starts takes place in mitochondria.
3. The cytoplasm then uses the genes in nucleus as program for cancer.
4. The program for cancer preexists in genes in nucleus, it only has to be activated and turned on.
5. The energy producing mechanism of cancer is fermentation. Fermentation takes place in cytoplasm; but not in the mitochondria.
Thanks Josh, I wanted to confirm to what extent we are on same page.
I guess point 4 is the area that needs more work. If these genes are pre existing from the time of low O2 atmosphere and single celled life, then what evolutionary force has caused them to be conserved; in other words why haven’t mutations rendered these genes non functioning? The only answer has to be that they are also used at some point in our development as multi celled organisms. If anyone can explain in mkre detail than this what use our body has for cancer genes, I would be grateful!
Hi Mark,
Used in first week of life. Early embryo is one solid mass of very fast growing cells. Looks and acts a lot more like cancer than normal differentiated tissue. Also embryo tend to replicate early forms. See early human embryo with gills and tail etc.
Life is like a hoarder, doesn’t like to throw anything out. Just ask anybody who almost died from ruptured appendicitis, why they thought having an appendix was a good idea.
That’s a good explanation Alan, I knew there had to be a reason we still have those genes. I wonder what would happen if we removed them with a (future) genetic therapy once we reached adulthood. Would there be a downside?
All organs have the potential for failure.
We understand the purpose of most organs so we do not routinely remove them simply because they may some day fail.
According to researchers at Midwestern University in the US state of Arizona, the appendix has a function.
I added a link below.
The tonsils were once routinely removed, but no longer because we now know the tonsils try to prevent germs from causing infections in other areas of your body.
The fact that tonsils or appendix become inflamed may be likened to a canary in a coal mine.
Just like cardiovascular disease, or liver failure, if you get to a doctor in time you can prevent mortal damage.
http://www.independent.co.uk/life-style/health-and-families/health-news/why-do-we-have-appendix-purpose-discovered-reason-scientists-does-gut-bacteria-midwestern-university-a7524086.html
Appendix:
In 2015 about 11.6 million cases of appendicitis, which resulted in 50,000 deaths. Leading cause acute abdomen, leading cause exploratory laparotomy.
Before modern surgery a major cause death .
Hi Mark,
What you call “cancer genes” I call, a program of how to survive in world without oxygen and without a blood supply.
Now consider the xygote, it has a food supply from egg, but no blood supply and no oxygen. It will take 1 week to make it through oviduct to implant in uterus where if will get blood supply and oxygen. So question for xygote is how to hold your breath for 1 week and at same time to grow very rapidly. The answer is the program for life before oxygen and before living things had a blood supply.. What you call cancer genes, the zygote calls a program for survival in first week. Note after implantation, at start of second week, the fetus begins to show organized growth.
Alan –
I want to clarify that this is Seyfried’s theory and Warburg’s, not mine. I don’t know enough to know whether I believe it, but I do think it deserves more notice.
Your points 1-3 are a good summary of the Warburg hypothesis.
Point 5 – that mitochondria aren’t involved in fermentation – is something that I missed, but I’ve checked it. Seyfried writes in response: “Mitochondria can ferment amino acids. Glutamine is the most abundant amino acid in the body. Glutamine is also the major fermentable fuel for cancer mitochondria, especially under hypoxic conditions. The mechanism for mitochondrial glutamine fermentation in cancer is discussed at length in our ‘Press-Pulse’ paper that was referenced in your piece.”
Point 4 is not something that I’ve read anywhere in my brief foray into the Metabolic Theory, but it makes sense to me. I think it is implied, and your point about cancerous metabolism and unrestrained reproduction being an ancient part of our evolutionary history is a good one.
– Josh
Thanks Josh,
Warburg was excellent scientist. The fact that to stay alive he had to be friends with Hitler should not taint his work
Here is a very comprehensive study on mitophagy and its possible role in cancer. ” Mitophagy programs…… Cell Mol Life Sci 2016;73 : 775-795.
Cancer is complicated.
Conceptually, a role of mitophagy in cancer is an intriguing hypothesis…..However,to date the roles for mitophagy programs in cancer remain unclear…..High Bnip3 is reported to correlate with invasive tumor behavior in breaststroke, colorectal,lung, prostate, and uterine…..Onthe other hand, it can be silenced in leukemia,pancreatic,gastric, and colorectal cancers.
Thus , these in Vito cancer studies suggest contradictory roles for mitophagy receptors and signaling regulators in cancer.
Cancer is an intelligent and muti headed beast and no one approach will master it I’m afraid.
What we really need to get at is what is the root cause of cancer? Or to put it another way, is there something causing those nuclear mutations other than just random, stochastic damage? I think it is reasonable to suppose that the metabolic dysfunction that accompanies aging, and which is exacerbated by the modern intake of calories could be at the root of this.
I think that a low NAD+:NADH ratio, which is what you get in a well fed state, might mean that the electron transport chain can’t keep up with the NADH input, with a resultant rise in ROS. This might not mean instant cancer, but it raises the odds over time. Couple that with immuno senescence and cancer is suddenly a much more likely possibility.
Dr. Dalgleish, the well known oncologist at St. George’s Hospital London, showed us a remarkable in house study of 20 cancer patients , each of them had evidence of chronic inflammation and a secondary ( literally approaching ZERO immune response system).
He primed these patients with metformin, ldn, alpha lipoic acid , prior to surgery, radiation, chemo., whatever the case may be. He got pretty impressive response rates.
But the ultimate determinant for their long term survival was their body’s ability to reduce the inflammation and bring the immune system back to a normal person’s state.They were the ones who made it.
Chronic inflammation and poor immune functioning are the constant bedfellows of cancer. LDN is the only drug that both reduces inflammation and modulates the immune system back to homeostasis.
So I am probably the only person in the world on both rapamycin weekly and LDN daily. But if you get a chance to stop by St George’s, treat Angus to lunch , and you’ll be the second person.
Hi Dr. Rivas,
I am a female, 54 years old. Since post menopause, my immune system has gone haywire. Any little change, temperature, air pressure, dust, spices, etc. induces allergic-like-symptoms. To the point, I can no longer live in Canada during the winter months. So I would definitely try ldn before rapamycin. What do you think?
Paul,
You are onto a very good regimen. Taming chronic inflammation and immunoscenescence gives us a fighting chance not only against cancer as we age but also against many other infections and diseases that kill us.
Hello Dr. Rivas, what Rapamycin dose would you recommend for ApoE4 carriers? Would 1mg / weekly probably be suitable, since its half life can go up to approx 110 hours?
Thanks!
Hi Mark
I sent you an email on multiple diet, foods and nutrients, ( but not supplements) , to lower inflammation from The Linus Pauling Institute. It also re-enforces your idea that our modern diet is killing us. I would have posted it here but couldn’t figure out how to do it!
Thanks Paul, I got your email this morning and will read it ASAP.
I can’t post the link either, but for others if you Google ‘The Linus Pauling Institute Inflammation’, it’s the top result.
This is a great overview, and it does seem to support my assertion that modern diet (i.e. excessive calories, particularly carbohydrates) contributes to cancer (via a rise in ROS and chronic inflammation).
There is also a brief section on vitamins and phytochemicals. It mentions B6, which is interest to me, as I’m very low in B6 due to a homologous SNP – and have found supplementing it markedly decreases my summer grass pollen allergy (B6 helps process histamine). Might be relevant to you Cassia if you have that SNP?
Hi Mark,
I don’t know if I have SNP. Have not been DNA sequenced. Mine allergies are probably due to some autoimmune disorder as I had Graves’ disease in my late 30’s. I do supplement with B complex and extra B12 (I am a vegan, occasionally eating salmon).
Hi Paul, would you be able to send the link to me as well?
Point 4 – and some of the other points above – seem like pages from Paul Davies’ theory of cancer. Physicists wading into biology tend to encounter a fair bit of push-back, rightly or wrongly. ; – )
Paul Davies is one of my favorite “deep thinkers” who has been bringing together ideas across disciplines for decades. His theory of cancer, in my opinion, misses the systemic picture. Of course, the cell has the ability to multiply out of control, which it inherited from the ancient past. But why does the body put up with it? Why doesn’t the body’s policing system (white blood cells) catch the offending tumor and destroy it?
My best guess is that proto-cancer arises often in healthy people, and that the cells succumb to apoptosis or they are handily eliminated. Cancer is a systemic disease that can’t occur in a healthy body with a healthy immune system. If my tumor is transplanted into your body, your body will reject it, and you won’t get cancer. Usually. In this sense, cancer is a disease of the whole body and not an isolated cell where something went wrong.
Of course, the Warburg-Seyfried theory is also subject to this criticism.
– Josh
Hi Josh,
These are 3 different stories:
One story is about the origin of cancer and how starts, the subject on this section.
The second story is the bodies defense, prevention of cancer from becoming clinical disease; this is story within anti-aging medicine.
The third story is the treatment of clinical cancer: this is medical field of oncology.
It would be fascinating to follow a group of people with no family history of cancer and very low inflammation and a fully healthy and functioning immune system c/w a similar group with a strong F.H. of cancer and see over a twenty year period if many, or even any, get cancer.
Hi Nick,
Exactly right. Major point, cancer from time when atmosphere lacked oxygen, straight from Paul Davies.
Idea that these genes support embryo very early period for rapid undifferentiated growth, Davies.
Connection that first week of life embryo lacks blood supply and oxygen so needs this program, my idea.
I tacked on a few wrinkles from Josh regarding role of mitochondria.
This theory provides no clues as regards treatment of cancer.
As regards prevention, suggests factors outside mere random mutations to genes in nucleus play role.
I wish you would not refer to environmental carcinogens or teratogens as “toxins.” Toxins are poisons made by living things such as rattlesnakes, jellyfish, bees etc. The environmental chemicals are toxicants, toxics, toxic chemicals etc. but are NOT toxins. The word is often misused in the popular press, but scientists should use words correctly. Thank you.
Gluconeogenesis keeps your blood glucose at around 4 mM (70mg/mL) even when starving for weeks. A good source for glucose is protein catabolism. If your blood glucose falls bellow 2 mM or 40 mg/mL, you die. Our brain cannot exist without glucose. I think thats is the reson why cancer cant be cured with dietary glucose deprivation
hi Gabor
That is true under ordinary conditions, but under starvation conditions the brain is very capable of living off of fat- derived ketones. Eating a bunch of sugar certainly doesn’t help your cause if you’re trying to prevent cancer, but neither will it cure it if you stop eating sugar.
Thanks. Still a short googling session led me to a paper that states that some colon cancer line can live off totally sugar free medium. so as you said cancer is much more complicated illness than it could just be reduced to “mitochondrial”.
anyways. total or near total glucose deprivation might still be the way to go. if the brain can somehow adjust to the lack of glucose, patients could be treated in deep sleep, slowly inhibiting gluconeogenesis and pushing down blood glucose levels. maybe cancer will become super vulnarable under these circumstances just as Longo’s research suggests.
Sugar is definitely the ‘best’ energy source, in that it is easiest to process for mitochondria, certainly much easier than long chain fats, which need more oxygen, produce more ROS and produce less ATP. So cutting out sugar will make it hard for the parts of the body that need lots of energy like the brain, and also cancer. So yes I agree with GaborB that a low carb diet would be helpful, but probably not a panacea.
Hi Mark
Even if you dramatically reduce your carb intake Cancer can and will outsmart you by using alternative methods of getting glucose.
Cell Metab 2012 Dec 5 16 (6) 751-64.
But one thing of interest is that cancer, in order to utilize glucose, stimulates and utilizes PARP14. So inhibiting that protein impairs the Warburg effect and starves th bastards to death. A promising intervention.
Parp14 promotes the Warburg effect……
Nature Commun. Iansante 10 Aug 2015
Very interesting, thanks Paul.
Hi Paul,
As you mentioned, PARP14 inhibition is currently investigated to fight cancer. This is just one paper (that you might already be aware of) but there are probably many others.
Very interesting. Thanks for bringing that to our attention.
Biochem Biophys Res Commun. 2017 May 6;486(3):626-631. doi: 10.1016/j.bbrc.2017.03.052. Epub 2017 Mar 14.
Identification of PARP14 inhibitors using novel methods for detecting auto-ribosylation.
hi Cassia
I’m not your doctor so I’m not in a position to give specific medical advice, but I can tell you that you have a well described problem and you’re not crazy. Allergies are one of the lesser known complications of menopause, and it is not caused by low estrogen per se , but by Fluctuating estrogen levels. You listed many of the common triggers.
Some people do get relief combining an antihistamine with flonase nasal spray and adding in ginseng with black cohosh, none of which require a prescription.Others require estrogen replacement to stop the fluctuations and your GYN can help with that decision.
hope that helps
I honestly don’t know about ldn and allergies, but it does make some sense, but I’m really not sure. Try contacting ldnresearchtrust.org for that one.
Thanks for your kind and prompt reply. I cannot tolerant any of the anti-histamines. Right now I am coping it by living in the tropicals where temperature and air pressure change the least. Will try out LDN soon and see how it works.
Thanks again.
If your doctor isn’t familiar with how to dose it properly let me know and I’ll be glad to speak with him.
Thanks for your kind offer. But I am not getting LDN through a doctor. When there is a universal care, it could also means no care (as in Canada). So I will do some online search and order it from somewhere. Most sellers do not ship to Canada. So when I say soon, it probably means 2 months from now.
Interesting read on alternative cancer treatment using old drugs.
http://virtualtrials.com/pdf2017/treatment_options_gbm_2017.pdf#page27
Thanks.
Hi Bill
Bogdanov wrote science fiction about Martian travel that used parabiosis.
Then tried it on himself and died of malaria.
Does you have a more convincing reference.
Hi Bill,
I had in mind pesticides, a common poison, causes aplastic anemia.
Autopsy and full toxicology very seldom done on old persons with chronic illness like unexplained anemia, so easy target for “perfect murder” until possibly the son-in-law mentions it on Josh’ blog.
Yes, Alan, you’ve outed my secret plot. Unfortunately that doesn’t explain why so many old people have low blood cell counts, they aren’t all out getting healthy agricultural labor and being sprayed with pesticides. Cell senescence would account for their condition, pesticides do not.
(And if they don’t do full autopsies on old millionaires where you live, I suggest you flee immediately 😉
I am fully aware of the Bogdanov story, including that it might just have been blood typing mismatch that actually killed him. Still, by all accounts he looked very healthy and energetic for a while on his stolen blood.
Are you pulling my leg about never having seen a paper on rodent parabiosis? They go back to ~1864 IIRC. Here’s a quick review article to get you started, if you’re serious:
https://www.nature.com/news/ageing-research-blood-to-blood-1.16762
Hi Bill, Thanks for great post and excellent review paper. You are very much one of my favorite contributors.
As regards prior post regarding repositioning old drugs, great note about angiotensin 2 blockers and anti-cancer effect. Angiotensin 2 blockers part of my anti-aging formula and excellent drug, does a lot more than lower blood pressure.
Quote from this paper as regards anti-aging drug, singles out rapamycin and says only drug shown to reliably slow or reverse aging across many mammalian tissue types, but neither [talking about CR] has turned into an anti-aging treatment…the latter (rapamycin) has toxic side effects”. Guess what, no toxic side -effects with weekly use and now rapamycin being used as cornerstone of treatment by physicians in office practice as anti-aging treatment. [at least one that I know of for sure]
As regards parabiosis, [conclusion]”For now, any claims that young blood or plasma will extend lifespan are false: the data are just not there.”
As regards the billionaire from Hong Kong who funded Alzheimer’s disease parabiosis study, no results yet.
However, I would like to do study to see if rapamycin will prevent Alzheimer’s disease if started early, before minimal cognitive impairment, in carriers of ApoE4 gene. If anybody knows this Hong Kong billionaire, please ask his people to call my people.
It would be fantastic if parabiosis works; my problem is with the entrepreneurs running way ahead of the data. No problem with researchers doing scientific study. As pointed out in prior post, because blood is legal product, there is nothing to stop physicians from using young blood transfusion for anti-aging.
Ah, here’s some more explanation of my weird confirmation bias about this 1864 idea. If you put fetal serum on BJ cells you can push another 20 PD out of them… young hormones definitely reset cells.
Obviously we need real controlled studies on both young plasma and actual connection of young immune systems to old. But dismissing it out of hand is not reasonable… we already know that you can transplant old organs into younger people (or organs from shorter-lived animals into people) and the younger blood resets the cells. The 1970s results showing 4-5 month life extension for rats has to be given some weight, too… those rats lived longer even under the stress of an 1860s procedure and being tied to another rat… that wasn’t even a clone!
Hopefully Alkahest will cough up some human trial data and we’ll see. Oh, and here’s a human-blood-in-mice paper:
https://directorsblog.nih.gov/2017/04/25/aging-research-plasma-protein-revitalizes-the-brain/
Hi Bill,
Now that liver comment.
We both agree can have 80 year old liver donor.
To me, nothing to do with being reset.
It means liver could perhaps live hundreds of years if wasn’t stuck inside an old person.
Hi Bill,
Rats study; would like to see actual study. Taking what you stated as true here is my interpretation.
Two rats being physically attached caused so much problems for old rat that not able to eat properly and lived longer due to CR.
Need to have control in which rats attached in same manner; but do not share blood supply, just physical attachment.
Without this type control, I would say has nothing to do with sharing blood supply.
Please post actual study in which increased life span in mice or rats, so can review entire study, see if valid control as to same physical connection but without blood supply connection. Also need to see actual study to be able to review what other experts said about study.
The critical thing is showing that connected blood supply and not physical body connection that makes the difference in lifespan.
Hi Bill,
NIH another great paper.
They used cord blood as in umbilical cord as in NEWBORN.
In Hong Kong supported Alzheimer’s disease study they used blood from men under age 30.
To me, blood in umbilical cord and blood from 30 year old adults is about as different as day from night.
In clinical medicine we have done hundreds of million of transfusions from adult to adults.
Nobody has every given anybody a transfusion of cord blood.
So if parabiosis experimenters are talking about cord blood, that is totally different.
Hi Bill Alan,
Great discussion about parabiosis. Bill, thanks for posting two great papers. Alan, thanks for your critical analysis of the papers. I have learned a great deal in parabiosis today.
Hi Alan,
I have found the following paper interesting. It is not really about parabiosis but it still falls in the category of transplanting cells from young donors to old donors:
“Transplantation of mesenchymal stem cells from young donors delays aging in mice”
One possible caveat is that mice in the control group seem to have a relatively short life span from what I know.
good point about cord blood being different from mere young blood… ask any cell culture guy about fetal serum vs. regular bovine serum.
Also good thought about possible CR in the para-rats… I don’t buy it for one second, but it needs a control for sure to prove that this was a real parabiosis effect. Too bad we suddenly stopped doing certain types of biology in the mid-1970s, we’d already know this stuff 😉
Hi Bill,
Thanks for comment.
I concede that fetal blood, cord blood, baby blood could have growth factors.
As regards blood, human donors must be legal adults, say 21 y/o.
What evidence is there that young adult humans have anything good in blood.
There is also Blagosklonny theory that age related disease due to Hyperfunction, which suggests young adult blood not helpful.
Josh,
In reference to your statement: “Medium chain saturated fatty acids like coconut oil”
Coconut oil is very high (perhaps more than half) in lauric acid (C12), which from what I understand does not act like a medium chain triglyceride (MCT) once inside the body.
Thus, while coconut oil is healthy, it does not provide the same benefits as true “biological” MCTs like C6, C8, and C10 oils, nor does it induce metabolic ketosis as efficiently.
Matt
Hi Matt,
Interested on your opinion medium length fatty acids as recently added goat cheese to diet. Aside from other considerations goat cheese is very good taste.
In contrast, I find coconut oil difficult to eat in any significant quantity. The taste is too strong for most recipes.
Josh, your opening statements are not correct. Stating that the theory that “Cancer is a genetic disease… …is more than 100 years old…. but it didn’t become the dominant view until the 1950s, when, after Watson and Crick, genes assumed an exalted position in the study of biology.”
DNA, the genetic code and its biological functions were completely unknown over 100 years ago so it is nonsensical to suggest that the theory that cancer was a genetic disease is ‘more than 100 years old’.
Anyone who has studied cancer biology will know that Nowell and Hungerford were the first to provide scientific evidence that that chromosomal abnormalities were associated with Chronic Myeloid Leukemia (CML) (Science 132:1497). Then in 1973, Janet Rowley showed that specific chromosomal ‘fusions’ were specifically associated with CML (Nature 243:290). Since 1973, more than 150 oncogenes (that cause cancer when mutated) and 100 tumor suppressor genes (that cause cancer following genetic inactivation) have been confirmed in many many many studies in both animals and humans. Most recently, genome-wide sequencing of large tumor cohorts further add to the simply overwhelming, reproducible and high-quality scientific evidence that genetic abnormalities underpin cancer.
So, maybe Josh made a couple of errors in his opening statements, but doesnt the rest make scientific sense?
The answer is No.
Overall the blog shows a poor understanding of the molecular basis of cancer biology.
Josh is right that cancer cells demonstrate the Warburg effect (where cancer cells upregulate glycolysis to fuel growth). But the existence of the Warburg effect in cancer in NO way suggests/proves/indicates/demonstrates that cancer is not a genetic disease. No credible scientist would ever make such a conclusion. It simply means that cancer cells utilize a very different metabolic fuel source that most normal non-malignant cells.
What about the nuclei swap experiments? Surely that proves what Josh believes in?
No. Again, the blog-a-sphere is full of people who try to use scientific data to prove what they want to prove. Its simply ‘information laundering’. A scientist knows that injecting the nucleus of a cancer cell into the cytoplasm of a normal cells results in a normal cells because of how the epigenome is re-written. It simply doesnt prove that cancer is not a genetic disease. Again, such proposals are only found in the blog-a-sphere because no credible scientist would ever make such a conclusion based on such an experiment.
Josh, my intention is not to attack you. I am simply arguing the scientific evidence. And it appears that you have sought out evidence to support your beliefs. Such an approach is flawed, unscientific and usually leads to inaccurate conclusions, which is the case in this occasion.
Efjay –
Thanks for keeping me honest, and for the opportunity to clarify.
First, I am not wedded to the metabolic theory of cancer. I don’t know enough to say whether it is ultimately correct, but I think I have seen enough to say that it deserves more attention, and that it offers new possibilities for cancer research that ought to be explored.
Second, genetics is older than DNA and the idea of heritability “factors” goes back to Mendel. Christofferson’s book traces proto-genetic theories of cancer back to the 18th century. In the 19th Century, Paul von Hansemann looked at tumor cells in a microscope and noticed that their chromosomes were distorted. Theodor Boveri [1914] usually gets credit for formulating the first comprehensive theory of cancer as a genetic disease.
As for the nuclear transplant experiments: I agree with you that epigenetic modifications are probably a crucial step in neoplastic conversion. The fact that a cancerous cytoplasm can induce such changes in a normal cell nucleus with un-mutated chromosomes is an important piece of the puzzle.
– Josh
Hi Efjay,
Great post. I remember when I attended med school 1963-1967 there was no clue what cancer was about; or just about anything else. My current impression is that outside of anatomy, I could just as well have gone to med school in the 19th century. Seems that in medicine, 1967 was much closer to 1900, than 2017, as regards understanding of basic science. Love your use of term “blogosphere”.
……and I love your idea that, ‘in medicine, 1967 was much closer to 1900, than 2017, as regards understanding of basic science.’
great insight!
Hi Josh
To the question is cancer a genetic disease or a metabolic disease , the answer is , Both. It’s a chicken or the egg type of question where it’s highly probable that certain metabolic abnormalities may cause the genetic mutations seen in cancer cells and mutations may lead to the metabolic abnormalities, and these may work together in a nasty synergistic circle.
Your last several posts have just re-enforced that notion in my mind.
The Haycock study showed a relationship between telomere lengths and cancer, and while I don’t agree with his conclusion, it is notable that the major increase in relative risk associated with telomeres , and DNA instability, was gliomas or brain cancer.
Your last post featured the theory and work of Thomas Seyfried. Almost all of his success involving metabolic interventions, such as the ketogenic diet are with , again, gliomas.
Yesterday Cassia posted a very comprehensive look at many metabolic interventions, as well as multiple supplements, which have been found to dramatically alter the course of gliomas.
Akshay, on his website , looks at many cancer preventing supplements, and the cancers which they prevent, and many times it’s also gliomas.
So I think that you have a great example here of a malignant cell type that is most likely an example of both problems operating at once.
Yes it does seem we are stuck in a chicken and egg situation, which is not surprising when we consider that we are made up of eukaryotic cells, and therefore are descended both from mitochondrial genes and the genes in the nucleus. These two systems are profoundly intertwined and thoroughly symbiotic, so we should expect that aging and cancer is neither entirely due to one or the other.
We have MTOR, telomere shortening and potentially other epigenetic changes driving aging from the nucleus and ROS and metabolic decline (probably) driven from the mitochondria. Inflammation seems to be at the confluence of both influences.
Cancer has to be part of this same aging process, as it increases so much with age. I agree with Josh that in a healthy body cancerous cells still appear, but either kill themselves (apoptosis) or are cleared up by the immune system before they can escape senescence. The king of apoptosis, p53, is down regulated by inflammation (body is trying to hold onto cells), and inflammation triggers an immune responses, which if chronic becomes exhausting. So the systemic rise in inflammation with aging is probably the main reason (diagnosed) cancer increases with age.
My mother died of cancer after years of problems with arthritis in her knees and ankles. If only I knew back then what I know now, I may have been able to save her.
Hi Mark,
Going along nicely until you put the cart before the horse. Inflammation doesn’t trigger immune response, immune response drives chronic inflammation. Cells participating in “inflammation” are not rogue actors behaving badly. They are foot soldiers following orders.
Best way to understand chronic inflammation is to look at atherosclerosis as atherosclerosis is best studied inflammatory process. The end result is not “exhaustion”; but rather organ damage as in arterial thrombosis, infarction and death.
The inflammation is driven by part of immune system, pattern receptors part of innate immune system and some believe what they are reacting to is oxidized lipoproteins they mistake as foreign invaders.
The most effective agent in preventing atherosclerosis leading to thrombosis and infarction is rapamycin which dials down the inflammation. The main way rapamycin is dialing down inflammation in this setting is slowing protein synthesis (all of the components of inflammatory process) by blocking mTOR.
The point is immune system drives chronic inflammation and frequently the innate immune system and pattern receptors are central. The term ‘chronic inflammation is not the Boogeyman; but it refers to a specific disease process going on in a specific location and each specific disease needs study to understand exactly what is happening.
Another example is Alzheimer’s disease; chronic inflammation plays major role in damage; but this is also secondary to immune receptors who mistake all the crap building up (amyloid deposits and tau deposits) as foreign invaders,
If you look at Blagosklonny’s recent post you’ll see that he mentions propranolol.
We had a prior discussion about the parasympathetic nervous system and the vagal nerve’s involvement in anti-inflammation.
It would seem,after researching this that the sympathetic nervous system, especially the beta adrenergic line, stimulates the cytokines IL 6 and IL8. Recent work at Hopkins demonstrated that when cancer cells accumulate and feel crowded they metastasize via IL6 and IL8 signaling. So one would suspect, as proof of concept, that beta blockade would reduce cancer risk, and indeed it does.
A large Taiwan epidemiology study showed up to a 50% reduction of multiple cancers using propranolol for at least 6 months!
It could well be that the parasympathetic system is cancer inhibitory. Sympathetic cancer promoting. All related to inflammation.
That’s interesting Alan, but I think rapamycin not only helps through a reduction in the proteins the immune system mistakenly attacks, but also through the reduction in the inflammatory signals sent out from senescent cells. This signaling is intended to help an area recover by spurring regeneration, and attracting innate immune cells to kill the senescent cells. But for some reason senescent cells accumulate faster than they are cleared, and this leads to a runaway effect of more senescent cells being created, more inflammation, more immune response, locked stem cells and exhaustion of somatic replacement cells. So I think this is yet another chicken and egg situation.
Hi Mark,
We are in general agreement. My point is hyper function is primary driving force and chronic inflammation is end result. So I see direction starting with hyper function, which increases senescent cells which drive inflammation through action of immune system.
Hi Paul that is interesting about beta blockers helping prevent cancer! Certainly fits in well as you say with our previous discussions on vagus nerve stimulation moderating inflammation. So i guess the key is to chill out!
Hi Mark,
Blagosklonny includes beta blockers in his anti aging formula (Koschei) as anti cancer drug.
I don’t include beta blockers. In my opinion a specialized drug to be used by cardiologist. Also I am very much into exercise and beta blockers are not going to help you make it up steep hill on bike when need all the heart rate you can produce.
The theory proposed by Thomas Seyfried might be provocative and not popular among experts, but if it was factually incorrect, he should not have been able to publish it in peer reviewed journals shouldn’t he?
In case someone has written a rebuttal of this theory, it would be useful to post the link here.
However, there are large prospective and retrospective studies showing that there is no reduction in mortality from cancer comparing hypertensive patients taking beta-blockers and nonhypertensive men. Such as:
1) “Survival in treated hypertension: follow up study after two decades,1998;
2) “Is Cancer Related to Hypertension or to Its Treatment,1998;
3) Medical Research Council trial of treatment of hypertension in older adults: principal results”,1992;
4) “Incidence of and mortality from cancer in hypertensive patients”, 1993
On the other hand cardiac Glycosides (ex.: digitalis like digoxin) have demonstrated not only properties of induction of apoptosis and inhibition of proliferation of cancer. Also a large reduction in mortality of cancer, in patients taking these drugs at low concentration doses.
Please see the following article
Carlos ETB Monteiro, “Cardiac Glycosides at Low Concentration Providing Neuro-Hormonal Effects.: The Final Solution Against Cancer?”. Positive Health Online, Edition 241, October, 2017 at http://www.positivehealth.com/article/cancer/cardiac-glycosides-at-low-concentration-providing-neurohormonal-effects-the-final-solution-against-c
Abstract
In a central article published in the present edition of this journal was introduced a new hypothesis postulating stress (chronic sympathetic dominance) as the inductive factor for the increased lactate production found in cancer patients.
In it has shown the important role of stress as the major risk factor for cancer, also discussing on how it develops lactate formation.
In the present article is postulated that cardiac glycosides (digoxin, etc..) at low concentration doses fit perfectly well with the hypothesis of stress as the primary risk factor for cancer being these fundamental drugs for its prevention and therapeutic.
The article also discusses laboratory experimentation and clinical studies using cardiac glycosides. These have shown properties of induction of apoptosis and inhibition of proliferation of cancer cells. This apart of a large reduction in mortality of cancer, in patients taking these drugs at low concentration doses.
It also tells that some cardiac glycosides have shown sympathetic and glycolysis (glucose consumption and lactate) inhibitory effects.
Finally, this article explore the role of endogenous digitalis-like compounds in cancer and in other diseases.
I have a friend in Ecuador who eats three leaves of Kalanchoe pinnata (Miracle leaf, air plant) a day. She told me that it was good for the throat. It contains cardiac glycosides as well. Study showed that it has anti-tumor activity as well. http://www.tandfonline.com/doi/abs/10.1271/bbb.65.947
Whenever I am in Ecuador, I eat one leaf a day. I think you need zone 9 or warmer to grow. Otherwise it is super easy, needs no care. Careful though!
Digitalis is extremely toxic drug. This not to say very valuable drug used by cardiologist for heart failure. However, trivial use of cardiac glycoside plants is medical insanity
Hi Carlos
Congratulations on your study. Great job and much to contemplate. Let me know if I have this straight. Cardiac glycosides inhibit:
Cancer proliferation
Sympathetic stress related catecholamines
Glycolysis
While promoting apoptosis.
And digitoxin is superior to digoxin
The lower the dose the better, or more precisely a serum level <1.
A marked reduction in cancer incidence over many years of usage.
I've probably given this drug to hundreds of patients in the past, but it's rarely used now it seems.
Is it safe to use as a prophylactic agent in those with no cardiac conditions?
One thing for sure, it was better tolerated than inderal as long as the blood levels were checked periodically.
I am always cautious of studies saying such and such a drug (I.e. beta blockers) have no benefit other than correcting some problem (I.e. hypertension). That’s like saying an anti aging therapy has no effect other than preventing aging. As we get older our blood pressure rises, and this needs to be corrected. I wouldn’t expect such a treatment to completely prevent cancer, but it certainly won’t hurt!
Of course nothing in what you have said proves mitochondria do not cause or contribute to the genesis of cancer.
Hi Ejfray,
Thanks for your comments. I have no expertise in cancer biology and I am certainly not in a position to argue in favor or against Thomas Seyfried’s theory. However, in one of the paper mentioned by Josh, he describes experiments about mitochondrial transfer to cancer cells:
“More recent mitochondrial transfer experiments support the general findings of the nuclear transfer experiments ( 50 , 51 ). The tumorigenic phenotype is suppressed when normal mitochondria are transferred to the tumor cell cytoplasm. On the other hand, the tumorigenic phenotype is enhanced when tumor mitochondria are transferred to a normal cell cytoplasm. These findings further suggest that tumorigenesis is dependent more on mitochondrial function than on the types of mutations in the nucleus”
Kaipparettu B.A.et al (2013)
“Crosstalk from non-cancerous mitochondria can inhibit tumor properties of metastatic cells by suppressing oncogenic pathways”
Elliott et al . ( 2012)
“Mitochondria organelle transplantation: introduction of normal epithelial mitochondria into human cancer cells inhibits proliferation and increases drug sensitivity
Breast Cancer Res. Treat.”
What is your interpretation of these experiments?
I wonder if the answer is that inflammation is causing epigenetic plasticity, with random genes being activated. Eventually with chronic inflammation an oncogene is turned on. These oncogenes then use mitochondria to feed themselves and spread, so replacing the mitochondria with fresh ones using the electron transport chain as they should in a non cancerous cell would impede the cancer.
You might be right and this suggest that mitochondrial transplantation might be a viable option for cancer treatments.
However, in the text above things are not explained as clearly as I would like them to be. For example if the author was saying that:
1) transplanting normal mitochondria into normal cells does not harm the cells (does not create any mutations in the nucleus)
2) transplanting cancerous mitochondria into normal cells turn them into cancerous cells (and does create mutations in the nucleus)
Then, this would suggest to me that mitochondria can play a causative role in cancer. However, the way it is explained in the paper is a bit more ambiguous than that.
I don’t think anyone knows for sure, but as we’ve been discussing above, it may well be a chicken and egg situation when either mitochondrial dysfunction can raise inflammation and make nuclear mutations more likely, or a mutation (or epimutation) can happen anyway, but then needs to manipulate mitochondrial function to spread the cancer. It’s a fascinating area ripe for further investigation.
Readers here want to know how to treat cancer: I suggest:
Read comments on Josh site
Then 4 years Med school
Then 5 year Oncology residency.
Then good understanding how to treat cancer.
Great article on Cancer as a Metabolic disease, thanks
Related to the present discussion there are some news. Follows the abstract of an article published this month that offers an alternative hypothesis linking stress inducing lactate as cause of cancer:
Carlos ETB Monteiro, “Stress as the Inductive Factor for Increased Lactate Production: The Evolutionary Path to Carcinogenesis”. Positive Health Online, Edition 241, October, 2017 at http://www.positivehealth.com/article/cancer/stress-inductive-factor-for-increased-lactate-production-evolutionary-path-to-carcinogenesis
Abstract
In the present paper is discussed about the recent evolution in the understanding of the role of lactate formation in promoting cancer.
On it is postulated the hypothesis that chronic stress is the major risk factor and inductor of the increased lactate production which might lead to the carcinogenic process. It also explains how stress develops lactate formation, what was discovered in 1925.
The current hypothesis support ketogenic diets for prevention and therapy for cancer. This inside the reasoning that while fats do not have appreciable effects on the sympathetic nervous system (SNS) or in lactate formation, high carbohydrate diets have significantly effects on both SNS and lactate formation.
At the end of the paper has a short explanation and link to a parallel article where is discussed cardiac glycosides (ex.: digitalis like digoxin) as the fundamental drugs for prevention and treatment of cancer.
For those interested there is a new study titled ” Lactate Metabolism in Human Lung Tumors” published yesterday with full access at http://www.cell.com/cell/fulltext/S0092-8674(17)31068-1
In it the researchers showed lactate is not only a waste product but also acts as a fuel source consumed by lung cancer cells growing in patients and mice
Nuclear transfer experiments show that some cancers could be caused by an epigenetic mutation, rather than a genetic one.
At least if we define ‘gene’ as a transcription unit of DNA.
This would still fit very well into Alan’s and Paul Davis’ theory that cancer is the result of the activation of a suppressed cellular proliferation programme.
There can still be oncogenes that when actually mutated contribute to the likelihood that this programme will be set off, and tumor suppressor genes that stop it from starting in the first place and that have been silenced.
This point of view would have a lot in common with the idea that ageing is not the result of a wear and tear stochastic process, but rather it is epigenetically controlled. It is not necessarily a paradigm shift and it does not contradict what we know so far, but it may point us in the right direction of where to look next.
Hi Jonathan
Give me your email and I’ll send you the post.
[email protected]
For those interested in research on ketogenic diets and cancer I recommend Csaba Toth et al’s work at http://www.paleomedicinia.com which has worked with over 4000 patients in Hungary.
Their paper Comment on ” Systematic Review: Isocaloric Ketogenic Dietary Regimes for Cancer Patients ” by Erickson et al outlines some of their findings.
https://www.researchgate.net/publication/318723756_Comment_on_Systematic_Review_Isocaloric_Ketogenic_Dietary_Regimes_for_Cancer_Patients_by_Erickson_et_al
Alan Green, follows the first quote from my article about cardiac glycosides and cancer that, apparently, you did not read:
“Although there is not total agreement on the nature and clinical significance of the effects of digitalis on the autonomic nervous system, the following points seem well established and generally accepted: I) the actions of digitalis on the autonomic nervous system are very important clinically and play a major role in determining the clinical pharmacodynamic effects of the drug; 2) with therapeutic concentrations of the drug, the predominant effect is activation of vagal tone; and 3) with toxic concentrations of the drug there may be activation of sympathetic tone.” August M. Watanabe, 1985 [1]
Hi Paul Rivas,
First of all thank you for words of support.
It seems that the benefits about digitalis on cancer were observed a long time ago. Please see at http://www.second-opinions.co.uk/heart_drugs.html#.WbVMyMZhXGj the following information:
Digitalis – the right Drug Used to Treat the wrong Disease – Cancer
“As a student at Purdue University in 1930, Wayne Martin had as a tutor a seventy-year old medical doctor who had lost his money in the 1929 stock market crash and had taken a $60.00 a month instructorship at the university. Using the files of the Indiana University School of Medicine this doctor had done a survey between 1900 and 1930 of patients maintained on digitalis for life (digitalis was then used for heart patients). What he found was that not one of them had died of cancer. He was unable to get his survey published. At Purdue, Wayne says, the staff thought him to be suffering from dementia and he soon learned that if he wanted to keep his instructorship it was best not to talk about digitalis and cancer (1).”
Paul, in general your understanding is precise.
However, digitoxin is not superior to digoxin. What happened was the wrong dosage of digoxin during many and many years in heart failure which was corrected during the last decade as mentioned in my article..
Regarding if it is safe to use as a prophylactic agent in those with no cardiac conditions, to my knowledge, there are no studies indicating the contrary when cardiac glycosides are given at low concentration.
Very interesting Carlos.
This sort of began with Mark referencing a study showing an anti-inflammatory role of the vagal nervous system. Studies have shown that the parasympathetic system inhibits various cytokines such as IL 6 and 8, both have now been shown at Hopkins to be key players in cancer metastasis.
The Taiwan study showed significant cancer inhibition with only the non-selective beta-blockers such as inderal and corgard, but not at all with any of the selective beta- blockers.
You have also pointed out that this can be achieved, along with notable other mechanisms as well , with digitalis.
We also know that behavioral mechanisms to increase vagal tone, like meditation and exercise, are also protective.
So there certainly does seem to a trend here.
Just as an observation, for many decades starting maybe in the 1960’s, a large percentage of elderly patients were on either digoxin or propanolol, and the cancer statistics were not as high as today when far fewer people are on either one them.
But I do find your work on digoxin and cancer to be fascinating to say the least.
Paul Rivas,
Indeed, there are studies (1) showing a pivotal role for the sympathetic nervous system and its neurotransmitters in regulating inflammation.
1) The Sympathetic Nervous Response in Inflammation. Georg Pongratz; Rainer H Straub Arthritis Res Ther. 2014;16(504) Full paper at http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4396833/
That is a really interesting paper! Need to give this more thought.
Carlos
It seems that the evidence is adding up.
I am wondering if anyone knows about hawthorn which appears to be an herbal equivalent of digitalis?
I have found 3 plant extracts that stimulate the vagal system ( valerian, black cohosh, and passionflower). All of which extended life span in yeast.
It certainly seems that the vagus nerve is a good intervention point against the stress/inflammation that is causing so much cancer in the modern age. The problem with natural products is that they always seem to do more than one thing, I.e. black cohosh boosting oestrogen. Hawthorne is an interesting one, I wonder what the best does would be?
I still think C60 has the most potential here, though understandably not many people want to put buckminsterfullerene into their body. This company is giving it to dogs. Check out livepetllc(dot)com and have a look at the effect of their C60 product on cytokines (under about us). Quite incredible!
Hi Mark,
Very interesting. I would really like to see more studies around C60. We need to go to the bottom of it.
Take a look at selfhacked.com for Joe Cohen’s review of C60. Some good and some bad
Yes definitely interesting. Of course, well designed mice life span studies are still missing. Hopefully we’ll get more data in 2018.
The page just seems like a brain dump, not all relevant. There is so little information on C60 in olive oil, which was the 1 astounding rat toxology and lifespan study. Many of the other studies are from C60 in water, or pure C60, which are completely different.
Kelsey Moody had some problems with C60 in olive oil from ready made suppliers, in terms of impurities, but he was dosing mice that had already been given cancer and had no immune system, and equating that with the C60.
I found hawthorn by Nature’s Way at vitacost.com for only $8/120 tabs at 80mg each which is a very low dose.
And you’re right, I won’t take cohosh and I was allergic to the passionflower, ( but I have all kinds of plant and grass allergies).
Aren’t they coming out with C60 data in January/
Hi Paul,
My understanding is that we should get C60 data in early 2018 but I am not sure about the month.
By the way, have you looked at the info about LDN on selfhacked?
Do you agree with what they say?
hi Aldebaran
That review was very comprehensive and detailed.
It’s amazing stuff isn’t it?
Just 2 days ago I spoke with Dr. Turel on the phone. He has enormous experience using it for MS patients while chief of neurology and has published his results.
We both agree that a 3 mg dose daily would be a good cancer preventive regimen.
We are also concerned that the recently approved drug Contrave has way too much naltrexone and that it may well be cancer promoting over time at those doses.
Yes I agree LDN looks amazing. If I had one of the diseases listed or a high risk of cancer, I would definitely try it. However, would you even recommend LDN for healthy adults without any particular sign of inflammation? Do you think it can increase life span / health span of otherwise healthy people? If yes, it could be a good idea to ask the ITP to try it on one of their mice cohorts. I remember rapamycin become really popular around 2010 after the ITP show a significant increase in mice life span.
Hi Aldebaran,
I suggest you read, “Low dose naltrexone. Bogus or cutting edge science” from Science based medicine. Also read some papers about LDN from people not part of the LDN community. You will find very little support for LDN outside of the LDN bloggers. Big difference between medical world and Blog world.
Hi Alan
I of course agree with your final sentiment but it’s not snake oil and there are multiple studies and anecdotal reports on its benefits from physicians.
On fibromyalgia out of Stanford
“LDN for the treatment of fibromyalgia” Arthritis and Rheum 65 (2) Feb 2013 529-538
Again Stanford LDN as a glial cell modulator.
” The use of ldn as a novel anti-inflammatory treatment for chronic pain” Clin Rheum 2014 33 (4) 451-459.
Remarkable Crohn”s response
Safety and Tolerability of LDN in children with moderate to severe crohn’s”
J Clin. Gastroenter 2013 Apr 47 (4) 339-45. Smith J P
Blocks cancer cell proliferation
” LDN targets the opiod growth factor receptor pathway to inhibit cell proliferation” Donahue
Exp. Biol. Med 2011 Sep 236 (9) 1036-50
And there are many more than those, not to mention numerous physician case studies of dramatic responses where they have no motivation to sensationalize it.
But it certainly doesn’t work for all disease states and the response rates are not 100%, and like all drugs one must consider benefit/risk. But I for one am not willing to dismiss it out of hand.
I can tell you of a little girl with autism who was virtually impossible to even communicate with a year ago, but now after one year on ldn she’s able to sit down calmly and do math problems.
I only bring this drug up for those on this site, not as an attempt to get them to take ldn, but rather to highlight it as a drug with very novel anti-inflammatory and immune- modulating effects where the basic science is quite sound.
But I would expect opposition. As Schopenhaer said, ” All truths pass through 3 stages, first ridicule, then violent opposition, and then acceptance as being self- evident all along”
I think that rapamycin is in a similar position don’t you?
It would be a fool’s errand to try to debunk every highly dubious claim put forth on this website.
However, I think readers should read paper “Low dose naltrexone Bogus or cutting edge science” and decide for themselves.
Spoiler alert: Criticism is directed not at LDN; but against the LDN community for grossly over promoting LDN out of all proportion to the actual hard core scientific evidence.
A good example is Aldebaron comment why not do ITP study and see how LDN promotes lifespan in mice. Exactly. That would mean more than 50 anecdotal stories. The LDN community has been promoting LDN for over 20 years; but nobody did a mouse lifespan study.
As regards your comment about rapamycin, there is no rapamycin society, no rapamycin convention, nobody with rapamycin blogs.
The comparison of rapamycin to LDN is as follows:
Rapamycin: evidence: promotion : 100:1
LDN: evidence: promotion: 1:100
The problem is the gross disparity between the quality of the scientific evidence and the degree of promotion by LDN community.
Alan
What do you make of this study suggesting that rapamycin used for a prolonged time actually reduces male lifespan.
Depletion of Rictor, an essential protein component of m TORC2, reduces male lifespan.
Dudley Lemming. Aging Cell 25 July 2014 911-917
Paul
It has ALWAYS been known that reduction of mTOR2 or Rictor is BAD.
The entire theory of use of rapamycin to prolong life was to reduce mTOR1 but not reduce mTOR2.
Transplant medicine aims to reduce mTOR2 Rictor to knock out immune system.
High dose rapamycin can knock out Rictor and have harmful effects.
This was genetic experiment in which showed females had normal life span but males short lifespan when knock out one gene and make heterozygous for Rictor. Study shows females higher tolerance for reduced level of Rictor.
Lamming has done very good job in showing the difference between mTOR1 and mTOR2.
However, your statement, “Rapamycin used for a prolonged time actually reduces male lifespan” is a TOTAL MISREPRESENTATION OF STUDY.
Coming immediately following my comment about the quality of scientific support for LDN, I suspect the intent of your statement to be malicious.
The study does not use rapamycin. It is a gene knockout study. It is not a rapamycin study.
Male mice fed oral rapamycin have 23% extension of lifespan.
I very much welcome the 2014 study showing danger of depletion of Rictor. Everybody using rapamycin must know that too much rapamycin would reduce Rictor which is bad. The trick is to reduce mTOR1; but not reduce mTOR2. this is achieved by intermittent dose.
Just wondering what is the exact reason that intermittent dose of rapamycin is set weekly rather than say every 8 days, 10 days or monthly. I understand that inhibition of mTORC1 is what we want for life extension. Since only chronic administration of rapamycin can inhibit mTORC2, therefore, intermittent dosing is chosen. But what is considered chronic and what is considered intermittent?
Hi Cassia,
You can do something daily, once a week, biweekly, monthly; but you can’t do something once every 8 days because no work in English language for once every 8 days.
I would probably hesitate recommending it for a healthy person, unless that person has a strong family history of an auto-immune illness or cancer, in which case one would have to evaluate the benefit / risk of that approach.
Hi Paul,
Thanks for your answer.
I read the article from Steven Novella that was recommended by Alan and I read your answer above. All of that is very useful information so thanks to both Alan and You for clarifying the picture about LDN.
I consider LDN as very promising for a number of diseases and supported by serious scientists but some bloggers have created too much hype around it. Still, some definite proofs are missing to convince the scientific community.
For me, in the absence of any particular disease, I am mostly interested to extend my (healthy) life span and to encourage other people to look at the research around anti-aging and make their own choice.
The main criteria I am looking for is evidence of robust life extension in mammals as well as support from serious scientists. It seems that we have that only for rapamycin and CR at this point. Both need to be carefully applied for human usage to avoid risks (weekly rapamycin and moderate CR).
Of course, healthy life style (regular exercise, healthy diet) is the very first step to adopt before anything else.
There is also evidence of robust life extension for 17 alpha-estradiol and acarbose in mice but the mechanism is unclear so I am on the sidelines. In addition, angiotensin II receptor antagonists looks quite promising but I am not sure if people with already low blood pressure should consider it.
I am certainly forgetting many stuff. I am not an expert, just an interested reader. My feeling is that the field of anti-aging has now reached a state where it has some real potential. Josh’s site is for me the most valuable source of knowledge I am aware of.
Here, here Alderbaran. Totally agree we are now reaching the point where – if we are clever and careful – we could put together a regime that could extend our lives significantly. Of course there are no guarantees, but I personally believe we are no longer just passengers hoping for good genetics or luck.
If have certainly got some ideas about substances I might try in the future, LDN is one of them. But for the time being rapamycin is the cornerstone of my efforts, along with high intensity interval training and occasional NAD+ boosting for mitophagy. I am sure that Josh’s blog will continue to be a brilliant resource for me to learn and safe guard my own long term health, and I am grateful to Josh and all commentators here for that opportunity.
hi Mark
You would really think that by now they would have at least repeated the rat study using the exact same C60 in the same exact oil, just with a bunch more rats.
Interesting review on a noxious weed, bidens pilosa:
https://www.hindawi.com/journals/ecam/2013/340215/
This plant seems to have anti-inflammatory, immuno-modulatory properties.
Hi Aldebaran
In terms of life extension I would certainly agree . There’s rapamycin and not much else, though C60 is intriguing.
Hi Alan
I wasn’t attempting to be malicious, That was the title of the Study, and since I’m male and on rapamycin it got me a bit concerned. But I asked your opinion because I’m not as familiar with the details of rapamycin as you are.
But I do appreciate the response and clarification.
You are a very kind person, Paul.
Paul:
Thank you for posting the information regarding the study
For those interested in the story of rapamycin and TOR discovery, I recommend the following paper:
“TOR, the Gateway to Cellular Metabolism, Cell Growth, and Disease”
Cell Volume 171, Issue 1, 21 September 2017
http://www.sciencedirect.com/science/article/pii/S0092867417309443
I note that the author is quite optimistic about the future of mTOR inhibitors:
“By identifying the rapamycin target, mTOR, and playing a major role in defining its function in cell signaling and disease, the legacy of Michael Hall’s original discovery is being further defined as mTOR inhibitors and inhibitors of other pathway components are winding their way through preclinical and clinical trials for cancer, diabetes, obesity, heart disease, neurodegenerative disorders, and aging”
I’m from Canada and would like to try out Rapamycin, can anyone recommend a source where I can buy it?
The AHA does not support the claim that coconut oil is healthy (at least, from the point of view of CVD) due to its impact on cholesterol level (raising HDL but also LDL). However, they do not take into account that coconut oil is rich in MCT (in particular lauric acid which is BTW considered as MCT according to wikipedia), so maybe they are not looking at the full picture.
“Dietary Fats and Cardiovascular Disease: A Presidential Advisory From the American Heart Association”
There is a section dedicated to coconut oil which start with the following:
“A recent survey reported that 72% of the American
public rated coconut oil as a “healthy food” compared
with 37% of nutritionists.94 This disconnect between lay
and expert opinion can be attributed to the marketing of
coconut oil in the popular press”
After the eye-opening study from Yale scientist Vishwa Deep Dixit which showed inflamed nerve linked macrophages hugging the sympathetic nerves in adipose tissue blocking messages from neurotransmitters and thereby not allowing the utilization of the visceral fat during energy demands is now followed by another study also exposing inflamed macrophage related dysfunction:
‘Sympathetic neuron associated macrophages contribute to obesity by importing and metabolizing norepinephrine’ by Ana Domingo et al Howard Hughes Medical Institute published in Nature Medicine 9th October 2017
One therapeutic strategy can be to target the
norepinephrine import and degradation enzyme and neuro-immunometabolic signalling or alternatively reverse the root cause in both the dysfunctions: chronic inflammation
Hi Akshay
This is about BROWN FAT IN MICE.
Humans do not have brown fat.
In mice they burn brown fat to stay warm.
This paper show macrophages can interfere with function of nerves. That is very interesting and may relate to nerve diseases like MS perhaps.
It has ZERO connection with obesity in humans. In humans visceral fat not under sympathetic control. Humans don’t burn brown fat to stay warm. We shiver and hair stands up to create dead air space.
This is another example of what I call junk science. One observation and then run with it.
Very nice finding, in MICE, sympathetic nerves use to burn brown fat. However, when apply to humans without scintilla of evidence has any relation to humans and obesity this is junk science.
Nothing on you, Akshay, and thanks for posting interesting paper.
Alan,
I know you mean well and are intensely allergic to junk science. Having said that apparently we do have brown fat behind the neck. This study doesn’t mention brown fat of mice but their white fat which turns to brown and leads to weightloss once the enzyme leading to import and degradation of norepinephrine was blocked. This is what the scientists involved in the study have to say “Genetic ablation of Slc6a2in SAMs increases brown adipose tissue (BAT) content, causes browning of white fat, increases thermogenesis, and leads to substantial and sustained weight loss in obese mice. We further show that this pathway is conserved, as human sympathetic ganglia also contain SAMs expressing the analogous molecular machinery for NE clearance, which thus constitutes a potential target for obesity treatment”
Mind you Alan this is said by Dr. Ana Domingo winner of Philanthropies Award of $650,000 given to 41 scientists from 16 countries selected to be the brightest emerging scientists by likes of HHMI, Bill and Melinda Gates Foundation, Welcome Trust and Colouste Gulbenkian Foundation. While giving her the reward they said “Ana Domingos is investigating new molecular strategies to fight obesity. She has discovered a direct link between fat tissue and neurons of the sympathetic nervous system, which plays a role in burning fat. Stimulating these neurons could one day lead to a new treatment to cause fat loss.”
They said “This is an outstanding group of scientists who will push biomedical research forward worldwide, and we are thrilled to support them alongside our philanthropic partners,”
She was selected from 1,400 leading scientists in the world.
This is her pedigree “Domingos is group leader of the Obesity laboratory at the Instituto Gulbenkian de Ciência since 2013. Before her PhD in neurobiology with Leslie Vosshal at the Rockefeller, Dr Ana Domingos studied mathematics at the University of Lisbon. At Rockefeller, she started her Obesity research career in 2006 as a postdoctoral associate of Jeffrey Friedman, who discovered the hormone Leptin. As a postdoc, Dr Ana Domingos used optogenetic tools to identify a neuronal circuit in the brain mediating the reward value of sugar. She discovered that Leptin has a regulatory effect on this circuit, influencing how much one likes sugar. In the fall of 2013, she started the Obesity lab, at the Instituto Gulbenkian de Ciência, in her home country Portugal. Domingos´ lab was the first to visualize the long-time conjectured peripheral neuron-adipose junctions in the adipose tissue. Furthermore, her lab demonstrated that localized activation of these peripheral neurons is sufficient for lipolysis and fat mass reduction. Thus direct and targeted pharmacologic activation of sympathetic inputs to adipose tissues could represent a novel strategy for the induction of fat loss and a new anti-obesity therapy that would circumvent the challenges of drug delivery to the brain. These findings were published in Cell, and were widely disseminated in Nature, Science as well as Cell Press. Dr Ana Domingos received international awards such as those given by The Human Frontiers Science Program and the European Molecular Biology Organization.”
Hi Akshay,
My problem is with the last 5 words in abstract, “potential target for obesity treatment”. Paper in Nature Medicine, which does not appear to be open access, so can’t read paper, only abstract.
Paper appears to be perfectly fine study about mice adipose tissue and thermogenesis and regulation by sympathetic nervous system and norepinephrine.
I appreciate your respect for my concern about junk science.
To me, junk science is when an author of paper makes a big leap beyond their finding in the study and speculates about application outside their actual finding.
Here the problem is jump from mice to human obesity. Perhaps if had the entire paper, the speculation about application to humans would be much more restrained.
Everything I know about visceral, midline obesity in humans, especially as applies to older people has nothing to do with thermogenesis and sympathetic nervous system control or brown fat-like issues.
My understanding is adipose tissue in mice and adipose tissue in humans is acting like two different organs. In mice, brown fat or white fat that mice can convert to brown fat is acting as temperature control organ under sympathetic nervous system control for thermogenesis. This is not the case in humans and does not appear to be the case in human central midline obesity.
This mouse theory of obesity challenges everything that I believe as constituting the basic understanding of obesity in humans. To demonstrate this application to humans would require very large number of studies in humans, clinical studies and basic science studies in humans.
However, since don’t have entire paper, perhaps word “potential” is used as anything is possible.
In summary, I have no issue with findings in paper as regards mice, it is the application to humans
Alan,
I for once agree with you that just because has been successful in mice does not mean it will automatically translate to humans. In fact as per the few really well researched articles on the web very few do. So yes they do not have any evidence with regards similar benefit in humans and till that time it is just speculation. What I found interesting is their finding that “We further show that this pathway is conserved, as human sympathetic ganglia also contain SAMs expressing the analogous molecular machinery for NE clearance, which thus constitutes a potential target for obesity treatment”
In the main study they must have ‘shown’ evidence for this through some experiments. Also I respect the education and career trajectory of Dr. Domingos – she has done pastoral with Friedman d8scoverer of Leptin. So given the above is promising enough for me to get excited that there is chance of either if the 2 studies I shared going through to human clinical trials in future as obesity is a major epidemic in our modern world. Another take away for me is that I personally do not need to wait for clinical trials as the underlying cause in both macrophage dysfunctions is chronic inflammation. I can take natural compounds which have evidence in trials to bring down rogue inflammation. Rapamycin too indirectly provides that benefit. So I thought the readers may find it interesting from that point of view.
Humans do not have large quantities of brown adipose tissue, but to say they have none at all is extreme.
The conventional wisdom was that humans are born with brown fat but this quickly dissipates as the child matures. But it now appears that some deposits of brown adipose tissue persist into adulthood.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2699856/
Hi Michael Askay:
Here is a link discussing “Anatomical Locations of Human Brown Adipose Tissue
Functional Relevance and Implications in Obesity and Type 2 Diabetes”
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3661606/
Thank you Heather
You are welcome Akshay.
Sorry I spelled your name incorrectly
I was recently struck by an article showing that the popular herb ashwagandha root extended the lifespan of C. elegans by an impressive 20%.
” Kumar, R . ” Withania somnifera root extract extends lifespan of C. elegans”. Annals Res Article.2013.
This was followed up with an article showing that ashwagandha also increases telomerase activity by 45%. ” W. somnifera root extract enhances telomerase activity in the human HeLa cell line. Raguraman,V. Advances in Bioscience and Biotech. Vol 07 (2016).
It is also known that Ashwagandha inhibits both of the inflammatories, NF-kB and COX.
This is also true of Milk Thistle which is also a telomerase activator. So I thought that this may be a common denominator, but I am unaware of any significant life extension from milk thistle.
But Ashwagandha has one other attribute in that it decelerates senescence, which milk thistle does not. ” Deceleration of senescence….. Widodo, N J Gerontol 2009 Oct 64 (10) 1031-8.
In fact if you look at the life extending substances, not healthspan but lifespan, they all seem to have actions on cellular senescence in common, not anti-inflammation or telomerase.
Rapamycin is a good example of the above.
White Willow Bark, recently shown to extend lifespan very significantly in yeast, is in fact an anti-inflammatory ( COX 1&2), but it does in fact up regulate AMPK and inhibits TOR, thereby affecting senescence, which is now probably its life extending effect.
Pietsch demonstrated a life extending effect of Quercetin in C. elegans, and studies have shown that to range from 16 to 23%. ” Quercetin mediated lifespan extension in C. elegans……” Biogerontology 2009 Oct. 10 (5) 564-78.
Quercetin is a senolytic agent as demonstrated by Zhu, Y Aging Cell 2015 Mar 9
The relationship with all of these agents on senescence rather than inflammation or telomerase activation , or even on combinations of the above, is rather surprising to me.
I would love to know what others think of this.
Interesting observations Paul. Senescence has been shown to cause inflammation in studies. A study that corrobates your comments ‘Cellular Senescence as the Causal Nexus of Aging’ by Csoka et al published 2016 in Frontiers in Genetics says “Thus senescence is a precondition for anatomical aging, and this explains why aging is a gradual process that remains largely invisible during most of its progression. The subcellular damage includes shortening of telomeres, damage to mitochondria, aneuploidy, and DNA double-strand breaks triggered by various genetic, epigenetic, and environmental factors. Damage pathways acting in isolation or in concert converge at the causal nexus of cellular senescence. In each species some types of damage can be more causative than in others and operate at a variable pace; for example, telomere erosion appears to be a primary cause in human cells, whereas activation of tumor suppressor genes is more causative in rodents. Such species-specific mechanisms indicate that despite different initial causes, most of aging is traced to a single convergent causal nexus: senescence. ”
Unity Bio a start up that has discovered a synthetic senolytic molecule raised $115 million from the likes of Jeff Bezos – one of the larger fund raises in anti-aging this year.
Hi Akshay
Very interesting study. What I was expecting when I looked at these lifespan extending substances was that they would have several things in common, i.e.,m TOR inhibition and telomerase activation, or anti-inflammation + a senescent cell effect, or perhaps they would have nothing at all in common and would all be acting through separate pathways.
Senescence seems to be the key target with each of them.
C 60 may be an exception but we really don’t know its mechanism yet.
Interesting about Bezos and Unity Bio. I certainly wish them luck.
Hi Paul,
This is certainly interesting. I am unaware of any life span study on mammals with “ashwagandha” but it would certainly be worth a try. Regarding Quercetin, however, it has been tried on mice by Spindler and al. and the result was negative (as with many other compounds supposedly good for health and longevity):
“Influence on Longevity of Blueberry, Cinnamon, Green and Black Tea, Pomegranate, Sesame, Curcumin, Morin, Pycnogenol, Quercetin, and Taxifolin Fed Iso-Calorically to Long-Lived, F1 Hybrid Mice”
Longevity experiments on C. Elegans are surely very useful but as you certainly know, two serious issues are sensitivity to genetic background and reproducilbility. For example:
“Impact of genetic background and experimental reproducibility on identifying chemical compounds with robust longevity effects”
Nature Communications 8, Article number: 14256 (2017)
I consider very unlikely that a compound which doesn’t extend the life span of mice can increase the life span in humans. However, I might be wrong.
This is certainly not to say that senolytics are not promising. From what I remember, it is rather that Quercetin itself is not a very efficient or very specific senolytic (at least in mammals). Maybe it needs to be combined with other compounds to be efficient.
Thanks Aldebaran
I wasn’t really aware of the serious limitations of C. elegans since it seems to be used so often. I’m also wondering if there may be synergistic positive effects if you took all of the substances that work on C elegans and give that concoction to a mammal. Synergy may be an effective solution, and of course it’s always difficult to get the dosing just right.
Hi Paul,
The lack of robustness of C. Elegans life span experiments has led to the creation of the CITP (C.Elegans Intervention Testing Program) by Gordon Lithgow and others to workaround the issues by testing compounds across multiple labs and multiple strains. So results generated by the CITP are likely more reliable than others.
I think your idea to explore synergistic effects of multiple compounds on mice life span is really good but I am not aware of any such experiment on mice (excepted Rapamycin + Metformin at the ITP).
However, the following paper did exactly that on C. Elegans with both good and surprising results so definitely something to explore:
Slowing ageing using drug synergy in C. elegans
Tesfahun Dessale, Krishna Chaithanya Batchu, Diogo Barardo, Li Fang Ng, Vanessa Yuk Man Lam, Markus R. Wenk, Nicholas S. Tolwinski, Jan Gruber
You teach me something new every day
Really glad that I can be teach you anything at all given how much I am learning from you and others!
Hey
Did you get a chance to read the study that you referenced above?
Pretty amazing
You mean the one about drug synergy in C. elegans? Yes I read it a couple of weeks ago. Very promising but this is only a first step in my mind. Next step should be to reproduce their results with various strains / labs (a la CITP). After that, run a mice/rats study with the most promising combinations.
I thought that it was interesting that two of the three drugs were antibiotics produced by the streptomyces bacteria . Rifampin has long been used as a tb drug and seems not to act as a CRmimetic.
Alllantoin is used in skin creams like Olay.
When used together with rapamycin , all 3 certainly gave an eye opening healthspan and lifespan extension. Literally close to a doubling which the authors claimed to have never seen before.
Certainly speaks to synergy, at least in C elegans.
Good points about allantoin and Rifampicin. They might be onto something new to slow down aging (outside of CR mimetics).
Also, they confirm life extension of some cocktails on flies, which is nice to see.
I definitely like their conclusion:
” Our proof-of-principle study suggests that
beneficial synergistic and additive interactions affecting key longevity pathways are unexpectedly
common and evolutionarily conserved. These data support the feasibility of targeting multiple
conserved ageing pathways using existing drugs to slow down biological ageing rate, an approach
that, if translatable to humans, would result in dramatic medical and economic benefits”
The life extension obtained on Flies with Rapamycin + Rifampicin + Allantoin looks quite impressive as well (76.6% mean LS and 76.6% max LS).
They had several interesting observations. One is that synergy is effective at sub-optimal doses of each substance. Full doses of rapa+metformin added no increase in life extension, but 1/2 doses of each did give a synergistic positive response.
The other important thing is that the greatest effect was if each substance worked through separate pathways.
I couldn’t find any longevity mechanism for rifampin except for a lecture given by a botanist on senescence, where he demonstrates that rifampin retards senescence on the leaves of certain plants. He also states that Meristem does not undergo senescence and is essentially immortal.
A similar finding was also seen with Allantoin, again with plants , in a paper entitled
” Method for protecting plants from stress and senescence”.
It’s interesting that inhibiting senescence keeps showing up, though I understand that plant studies are a stretch, but still that seems to be the common goal of all of these life extending agents, albeit by different pathways.
Interesting findings about senescence. On my side, I have found 2 articles of interest about Rifampicin but they seem unrelated to senescence inhibition AFAIK.
The first one is about C.Elegans again. They claim that RIF extends life span of C.Elegans through AGE inhibition. If true, this could have big implications for diabetics (and even for non-diabetics).
“Rifampicin reduces advanced glycation end products and activates DAF-16 to increase lifespan in Caenorhabditis elegans”
Aging cell Volume 14, Issue 3 June 2015
The second article focus on the anti-inflammatory action of\ RIF on neurons.
However, it is not open access so I could only read the abstract:
“Rifampicin inhibits microglial inflammation and improves neuron survival against inflammation”
Brain Research
Volume 1395, 13 June 2011, Pages 12-20
In any case, RIF looks more and more interesting for anti-aging. I hope mice studies are ongoing.
I find the combo of rifampin/ rapa/ allantoin to have pretty exciting potential. It’s very interesting to me that both rif and rap come from streptomyces bacteria. The major side effect from rif was 5/850 people got liver toxicity. They may already have had some liver damage from other causes like alcohol, the study didn’t specify.
You can get allantoin off of Amazon in a powder.
Can you imagine adding a little C60 to the mix and getting 4 anti-aging agents all acting via unique pathways in a synergistic fashion.
Could be good!
Seems that RIF side effects are overall pretty acceptable. Thanks for the info. Very exiting indeed.
Here is an interesting article regarding cellular senescence
https://www.sciencedaily.com/releases/2017/03/170308092443.htm
From the Article:
[ They discovered that the enzyme SETD8 methyltransferase, which adds methylation on histone H4 lysine 20 (H4K20), regulates senescent features.
Normal cells stop proliferation after dividing many times (replicative senescence), and when oncogenes are activated for cancer initiation, senescence occurs to prevent it (oncogene induced senescence).
the past, SETD8 was reported to regulate cell proliferation and gene function via H4K20 mono-methylation, but its relation to cellular senescence was unknown.
The researchers, however, found that SETD8 decreased markedly in senescent cells.
When they performed a gene knockdown experiment (using RNA interference) to suppress the function of SETD8 in fibroblasts, cellular senescence was induced with typical features.
Furthermore, using a drug that inhibits the enzyme activity of SETD8, similar senescent cells appeared.
In other words, SETD8 plays a role in preventing cellular senescence.]
The article conclusion:
[ This research reveals that SETD8 protects against cellular senescence. It is expected that this result will be useful for understanding the mechanisms of senescence and developing a way to control cell aging.
This finding was first reported in Cell Reports on February 28th, 2017. ]
Interesting findings aboout SETD8. I would be curious to see if SETD8 reduction or over-expression have any impact on animal life-spans.
One cautious note about the article is that they seem to overstate the importance of senescent cells for aging:
“Since senescent cells are more active than expected, cellular senescence is considered to be the cause of the aging phenomena for the whole body. For example, it has been reported that when senescent cells of old mice are eliminated, whole body aging can be suppressed. In other words, if we can adjust cellular senescence, whole body aging may be controlled.”
Suppression of senescent cells has so far produced very interesting results on mice but the magnitude of the increase in maximal life span increase is around 10% (recent talk from Judith Campisi) , though effect on health span and mean LS is more impressive. This suggests to me there are more important factors driving aging.
Hi Aldebaran,
Senescent cells are leading actor in Blagosklonny theory of aging due to Hyperfunction. Number one bad thing mTOR doing is turning quienscent harmless cell into senescent cells which become large Hyperfunctioning and do very bad stuff.
I read paper and agreed with their characterization of harm senescent cells.
Now in late aging, there may be other major things; but when talking age related diseases in 65-95 age group I think senescent cells
major. At present, best way to prevent senescent cells formation is reduce mTOR. As regards Judith Campesi, hard to predict what will happen especially when involves the future.
Not sure what she bases those comments on since rapamycin through TOR inhibition suppresses senescent cells and leads to more like a 25 % increase in max life span.
Hi Paul,
I saw Campesi comment only need take once every few years. This was my interpretation.
1. It will be so expensive, only to afford once e dry few years.
2. It will make you so sick, will take a few years to forget how sick you were.
We will see when have drug on market.
Hi Alan
I feel that for all of her expertise in the matter, that she may underestimate the importance of senescence as it relates to both aging and age- related disease.
Hi Alan, Paul,
Thanks for your comments about the importance of senescent cells in aging. Always great to have your opinion.
The comments from Judith Campisi about max life span can be seen here around min 17 in the following talk from:
“Prolonging Life Span? – Judith Campisi – Rejuvenation Biotechnology 2016”
To my knowledge, the max LS extension obtained with rapamycin + metformin on mice is around 10% on male and 17% on females (at least at the ITP):
“Longer lifespan in male mice treated with a weakly estrogenic agonist, an antioxidant, an α-glucosidase inhibitor or a Nrf2-inducer”
Finally, I want to mention a recent TED talk from Andrei Gudkov (from Everon bioscience) that was posted here recently by John: “Can we stop aging?”
https://www.youtube.com/watch?v=L3irS84U8rs
His point is that senescent cells are rather the tip of the iceberg (around 12:30) but there is a more fundamental epigenetic aging clock which drives cells to become senescent with age (driven by retro-transposition).
I don’t have a strong opinion here but just want to provide information for more discussions.
Hi Alan,
I certainly agree that, as of today, the best anti-aging strategy is to reduce mTOR (through rapamycin, CR, exercice in particular).
Hi Alan,
Regarding the theory of Blagosklonny, my understanding is that TOR become hyperactive with age and drive cells to become senescent. Therefore, reducing TOR reduces the production of senescent cells and likely reduce the rate of aging. This is what rapamycin does.
What is not completely clear to me is the following: how much senotherapy (the removal of senescent cells resistant to apoptosis) will affect the rate of aging.
My understanding is that reducing the production of senescent cells by reducing TOR and removing senescent cell with senotherapy are two different ways to address aging. I know that Blagosklonny address the first point but I do not know if he address the second one as well.
In the article above, my understanding was that the authors were saying that clearing senescent cells (not reducing their production through mTOR reduction) would control whole body aging. I thought that was a bit of an overstatement as of today. However, I might have misunderstood their statement.
To be clear, I think senotherapy are one of the most promising future anti-aging treatments but I don’t know how much we can expect from it in term of max life span increase. You have certainly better information than me about that.
Hi Aldebaran,
Maximum lifespan increase (105) would take major advances in understanding aging PLUS major discoveries in treating those new discoveries. My guess is that is beyond the lifespan of anybody now alive.
There is what Blagosklonny calls aging (and age-related disease) and what Blagosklonny calls post aging syndrome. Post aging syndrome is about why people die rather quickly after age 100. Nobody has any real idea about that age group and why that happens.
Removal senescent cells in same category as rapamycin and addresses age-related diseases in pre 100 age group. It would be excellent if it worked. My guess is would be too toxic.
So best can hope for at this time and with what on the horizon is adding 20 years to health span and maybe few years to lifespan. Nobody is getting out alive,
Alan,
Thanks for clarifications about Blagosklonny theory and your perspective about clearing senescent cells.
My sense is that we are making huge progress in our ability to collect the data that could be relevant for aging (genome, epigenome, metabolome, microbiome). The only think I am confident about is that this technological progress will continue in the 2-3 decades to come. It is tempting to bet that this will help us to better understand aging in the near future. However, this is just my speculation.
Hi Aldebaran
I think that we are making huge strides in extending maximum lifespan. In from rapa logs to anti-aging formula Blagosklonny states ” Given that rapamycin consistently extends maximal lifespan in mice, rapamycin will likely allow mankind to beat the current record of human longevity, which is 122 years.”
We’ve also seen how some experimental agents like C60 and the combination of rapamycin with rifampin and allantoin show great promise.
The judicious use of senolytics may add even more.
I think that utilizing synergy in attacking multiple aging pathways may be right around the corner, and will make an enormous impact.
Hi Paul,
I agree with all of what you say. It is great to see that some experts are optimistic about delaying aging (maybe even more than what we have today with rapamycin).
One of the main question is: can we do something about the “Post aging syndrome” in the not too distant future.
In addition to all the things you have mentioned I am wondering if epigenetic partial reprogramming could be another viable option.
I am optimistic that the record of 122 will be broken but if it is only due to rapamycin, we’ll have to wait at least 40-50 years to see that. Best case: a “natural supercentenarian” (someone who would reach 110 without anti-aging treatment) currently in his 70-80 years old is on rapamycin today and he’ll delayed his death by 5-10 more years to break the record. And I think the chance of living to 110 today are 1 over 10 millions or so.
I agree. When you say epigenetic reprogramming, are you thinking of an environmental manipulation or a genetic one, or both? And how would that be done in your estimation?
Probably referring to the changes that occur as we age in the epigenome which alter gene expression and repression. There are multiple approaches currently to reverse some of the detrimental changes. For example everyday some cells turn cancerous but one of ways they get eliminated by apoptosis is via Tumor Suppressor Genes. As we age through epigenetic changes Tumor Suppressor Genes are silenced by methylation of their promoter. It has recently been found that some Cardiac Glycosides may reverse this silencing thereby upregulating TSG expression this was discovered by Jean-Pierre Issa and his team at Temple University. Other scientists are discovering other approaches to reverse adverse epigenetic changes.
Thanks.
Then would IP 6 have a similar effect by upregulating p53?
Yes
Has IP-6 produced any life extension an an animal model? Could be promising.
The problem with Shamsuddin MD PHD, is that his sole interest is in disease treatment and prevention, but not with life extension, so as far as I know, nothing has been done in that arena.
In his book entitled simply IP6 he does show through multiple in vitro and in vivo mouse experiments the incredible ability of IP6 to prevent all age related diseases, including cancer, CVD, dementia, osteoporosis, and Parkinson’s.
I only know of rapamycin that hits them all like IP6, but through an entirely different mechanism, of course.
I’d strongly recommend his book to anyone interested in preventing ARD’s.
Hi Paul,
Thanks for the info about IP 6.
Just started to read:
“IP6 in Cancer Therapy: Past, Present and Future”
Agata Matejuk and Abulkalam Shamsuddin
My first impression is that it certainly look promising but since IP 6 is already present in appreciable quantity in grains cannot we have the same benefit by eating lots of grains? Probably the answer is in the paper.
Also, pilot studies for cancer were promising but has there been more cancer trials since 2010?
It would be useful in my opinion to start a mice life span study with IP 6. I understand that it is expensive and that Dr Shamsuddin might not be interested that much though.
It seems IP6 obtained from food is bound to protein. Before it can be absorbed by the body, it must be freed from this protein by the enzyme phytase that is present in food and naturally in the intestinal tract. The power of the phytase enzyme is damaging to IP6 and renders much of it inactive and therefore less effective when obtained in this form. Pure IP6 from a supplement is not bound to protein and is easily absorbed intact and able to provide its complete medicinal properties.
Hi Aldebaran
Akshay hit on it with his response that for once a dietary supplement is actually more bioavailable than the food source., but in areas of the world where corn is the staple in their diet, the cancer incidence is quite low, but it takes alot of corn to get the protection. Shamsuddin outlines in his book a whole series of experiments that were done to prove that IP6 was responsible for the protective effects.
It’s really too bad that clinical trials on humans are so expensive. No great monetary incentives with supplements.
Hi Akshay,
Thanks for these precisions!
Hi Paul,
Thanks.
I understand that many promising compounds are not tested in clinical trials because of cost. Too bad but it is a reality. In term of brand, would you tend to recommend a particular one for IP 6 supplement or any would be OK.
Thanks again
Hi
I get IP 6 from vitacost and then I order lnositol separately in a powder from bulk supplements. The main thing is that you want a 4/1 ratio of IP 6 to inositol for maximal effect.
Thanks Paul,
This is very helpful information!
Hi Paul,
I was thinking in particular about the kind of approaches described in the following papers (which unfortunately are not open access):
“In Vivo Amelioration of Age-Associated Hallmarks by Partial Reprogramming” Cell Volume 167, Issue 7, 15 December 2016
“Anti-Aging Strategies Based on Cellular Reprogramming”
Trends in Molecular Medicine
Volume 22, Issue 8, August 2016
However, this is very recent so we’ll have to wait few years to see first results in normal mice (so far they have demonstrated the concept on a mouse model of premature aging).
Do you think it looks promising?
Thanks
I’ll check them out!
Hi Aldebaran,
I think there is one case of eigenetic reprogramming. The case of oldest living man in world at 113, excellent mental shape who was rescued from Aushwitz in Jan 1945, age 40, survived 6 months, 80 pounds when rescued by Russians. I think 6 months hard labor and starvation did something.
Hi Alan,
I agree. Very interesting case. Looks like there could be some potential if we find a safe way to apply epigenetic reprogramming to humans.
Just saw he died August 1 month before 114 birthday.
Hi,
It also occured to me that holocaust and world war survivors live so much longer than average people. But it could just be selection bias. They may have survived these hardships because they were more fit than their peers. And they lived longer for the same reason.
And there was Mouse treated 3 months with Rapa and then lived equivalent of 150 year old human. I don’t think conclusion was this was one fit mouse. A lot of people are fit. The chance this survivor oldest living man world is about 1 in a billion.
I’ve thought about this before. I wonder if suppressing MTOR to a very low level for 6 months can permanently suppress MTOR even if eating habits return to normal? We would need to know the details of his life after he regained his freedom. And as Gabor said, we’d not know for sure he wasn’t born with a low MTOR.
I have always read that in order to get longevity benefit for CR one has to get 100% RDA of vitamins and micro-nutrients but this case seems to show that CR (or mTOR reduction) is more robust than that. Clearly, they were malnourished in camps.
Hi Paul,
I was not aware of the controversy about PR. Thanks for posting data.
The only way I can think of to drastically reduce MET intake would be to eat only proteins as supplements from individual amino acids. Clearly not practical for human (unless you have lots of time to spend, lot of will, and lots of faith in the result).
On the other hand, you can moderately reduce MET by simply privileging food with lower MET % (ex: almonds). Josh posted a list of such foods a while ago. The question is does it help or not. I think there is a controversy about the fact that you need to reach a certain threshold of restriction to get any benefit at all. So I really don’t know if it could help to moderately reduce MET intake in this way.
Hi Aldebaran,
Remarkable that some people will write entire papers about methionine and never mention mTOR.
Quote from Blagosklonny; If you don’t know anything about evolution , you don’t know anything about biology and if don’t know anything about mTOR, don’t know anything about aging. (Paraphrased)
So low methionine means less stimulation mTOR.
Hi Alan
But wouldn’t low protein in general then lead to lower TOR? Why methionine in particular?
Hi Paul,
Not all amino acids equal.
Leucine high in milk more stimulation mTOR and methionine high level stimulation.
Vegan diet low methionine
A severe MR doesn’t seem practical. Some now think that most of the benefits the Mediterranean diet are due to the relatively low protein content of fish. Other data are now showing a protective role of animal proteins in preventing osteoporosis. Even with CR, is it the low carbs, low proteins,low calories?
Is any of it at all clear?
Hi Alan,
Well I guess it depends of the date of the publication. That’s probably understandable before 2006 but in 2017, that would be surprising.
I have found the following short review very readable and informative about epigenetic reprogramming:
“Bursts of Reprogramming: A Path to Extend Lifespan?”
Salah Mahmoudi and Anne Brunet
https://web.stanford.edu/group/brunet/Mahmoudi%20and%20Brunet,%202016.pdf
Yes, Belmonte et al expressed the 4 factors (called OSKM for short) required to create induced pluripotent stem cells (iPSCs) in progeria mice, but only for a short time so that they didn’t actually create iPSCs randomly in their bodies (this caused cancer) but just caused the cells move back towards being younger and rejuvenated the mice. One of the factors c-Myc (incidentally an oncogene), is right next to HTERT so telomeres were extended, and the other factors actually turned back the Horvath clock in these cells. So this process has incredible potential.
I am not sure whether we need to do more than just re-lengthen telomeres as this resets the most important gene expression. At this point we don’t know what benefit resetting the rest of the methylated or de-methylated gene expression is giving us, but I would put that speculation firmly in the post-aging syndrome box – I guess we will find out when we have fully dealt with cellular senescence, which has nothing to do with the epigenetic Horvath clock. But that notwithstanding this is the way to go to undo the damage done by MTOR and mtROS, which are driving aging, and which we can currently only slow, not reverse. So yes, very exciting!
On the importance of cellular senescence: I regard cellular senescence as being aging, as we currently experience it (notwithstanding other mechanisms that may kick in later). I will explain my reasons for this opinion. Cells want to grow and divide. This is the state of youth. Even if they are quiescent for a time, young cells have the capability to divide if required. Senescence stops all that, so you have no ability to bounce back and regenerate if required to. The problem with enhancing things like p53 is that this is the choker that holds back youthful growing cells from being young and exuberant and maybe forming cancer. So it is a balancing act. More p53 actually leads to more cellular senescence and more aging. But less p53 leads to a greater chance of cancer. (Additionally more senescence can lead to more cancer in the long run). So we absolutely need to remove senescent cells. Campisi and others are doing this now in clinical trials and it will be the next big thing in medicine. Of course this will only very slightly reduce the rate of aging due to reducing inflammation, you will continue to accumulate more senescent cells and will need to be treated again at some point. And Alan is right that this could be dangerous in the elderly – but not just because of the effects of the treatment (new approaches that are far gentler are in development), but because stem cells will need to be mobilized to replace the aged (senescent) cells, and the very old may not have this capacity. So you can see why max lifespan is not pushed out that far simply by removing senescent cells.
Hi Mark,
Thanks for your comments.
I agree that transient epigenetic reprogramming using the 4 Yamanaka factors looks very exiting and full of potential. This also suggest that epigenetic changes might play an important role in the aging process. Maybe, as you said, we’ll even be able to address the post-aging syndrome with this sort of approach.
However, this is still very very early and we’ll need to wait for more results to get a better idea (in particular, can it increase the life span of normal mice?).
I am also wondering about the possible risk of cancer associated with the 4 Yamanaka factors. The limit between effective reprogramming and risk of cancer might be tight. Hopefully, they’ll be able to figure out a safer approach.
It is tempting to think that epigenetic changes could be the leading cause of mTOR hyperactivation with age but I have not found any paper to back this up so maybe this is just wrong. On the other hand, I am not sure what other molecular changes (than epigenetic ones) could drive that.
Because epigentics is at the forefront of aging research I think there is a tendency to think epigenetics is something magical and must explain everything. But it is just different genes being expressed or not.
MTOR drives insulin resistance, so insulin goes higher, driving MTOR higher too. Absolutely epigenetics will be involved in this, but I don’t think it is causal necessarily. I think it more likely that a higher MTOR just accelerates the pace of epigenetic change, as measured by the Horvath clock and probably accelerates telomere loss as well.
These changes are essentially irreversible without telomere elongation or yamanaka factors, so with you I will watch the work of Blasco and Belmonte over the next few years with great interest.
I agree with you that it is likely that MTOR causes some epigenetic changes since, as you mention, higher MTOR correlates with the acceleration of Horvath clock.
On the other hand, I am wondering what difference between an old cell and a young cell can explain that the old cell has a more active MTOR than the young one under the same conditions. I have not found any answer yet in the literature, but it seems plausible to me that the accumulation of epigenetic changes is at least partly responsible for that difference in behavior.
Just as you, I find the recent development around partial epigenetic reprogramming fascinating because it might lead to a better understanding of aging and to more efficient treatments (although I am not sure about the time-scale).
Hi Aldebaran
According to Blagosklonny, the program remains the same. The cell changes. The program doesn’t change. The Blagosklonny theory explains why same program has different effect in same cell at different times. It may be that epigenetic changes are the tail of the dog and the dog wages the tail, the tail does not wag the dog.
Hi Alan
Even if epigenetic changes are not the cause of aging, they could still be a way to treat aging.
Check back in 22nd century.
This is absolutely my view on aging too Alan. MTOR is the driver (probably along with MtROS), of aging (including epigenetic change) and reducing MTOR slows aging. But to reverse aging we’ll need to actually reverse the changes aging has caused, like epigenetic changes.
I’m a little more optimistic than you, I don’t think the FDA and other regulatory bodies will have any choice but to change if they don’t want every country on earth populated almost entirely with old people who are not working. And even if they don’t the technology is changing so fast that this will soon be completely out of their control. But taking rapamycin is a good insurance policy to make sure we live to see that day.
Hi Mark,
Politically big push will not be productive old people. FDA and politics will be oppose anti-aging and any increase in lifespan. As increase health span could lead to increase lifespan; government will be very suspicious of increase healthspan.
Other countries may be different; but I’m sure in U.S. Right wing which runs country will very much oppose anti-aging.
I need to ask you a question Alan on rapamycin treatment please.
How important do you regard controlling the glucose increase in the blood that may accompany intermittent rapamycin usage? I know this is generally a benign effect of the body saving sugar for the brain, but whilst I initially dropped my Berberine usuage I am now thinking that was a mistake.
Best Regards, M
Hi Mark
Only risk is non enzymatic glycation. Opposed by metformin and use leg muscles ( walking) to promote insulin sensitivity. Very complicated discussed Koschei the immortal by Blagoskllonny.
I’m not sure that is true Alan. Governments are at least paying lip service to extending healthspan, and reducing the burden on health services from things like Alzheimer’s.
Plus rich old people want to live longer, so I can see support from the Right on this too.
But I take your point, there will be a lot of resistance from some quarters. My prediction: initial resistance will suddenly flip to wholesale support. but it’s anyone’s guess when this will happen.
Hi Alan,
Like your humor!
Maybe you are right about the 22th century. It is impossible to say.
However, nobody could have predicted the discovery of IPSC or the CRISP technology back in the 20th century. Nobody would have predicted around 2000 that we would be able to do full genome sequencing for less than 1000$ in 2017. Experts in AI were wrong to predict in 2014 that computers will not beat the world champion at GO before 2025. So maybe some major discovery about aging are lying ahead and we have no clue.
On the other hand, I am sure that back in 1970, everyone was convinced that we would travel to Mars long before 2017.
I think that if any country in the world develop anti-aging to a point where its population live significantly longer, US will follow soon. They don’t like to be behind others when it comes to technology.
Hi Alan, Mark,
My view of aging is that I don’t understand it. I am really impressed by Blagosklonny because, in particular, he was able to predict that rapamycin would delay aging before experiments on mice confirmed it. And his theory seems to fit very well with current facts. However, as far as I can tell, he does not say much about how the aging quasi-program is implemented in our cells or in our body. He describes very well the effects of the quasi-program though, but not its precise mechanism. So it seems to me that some progress could be made on that point. However, it could be that I am completely missing something.
Hi Aldebaran,
Reread 2006 first Blago paper Immortality and Quasiprogram. Nowadays people just use term antagonistic pleiotrophy for same concept. It is implemented in cell as same program runs cell from birth to death.
During growth and development mTOR program runs very well. When cells stop cycling they need different program .
Reread Blago , Speeding Car.
Old cells need a fix, a patch, a computer update.
Rapa is attemp to provide fix for mTOR program that works so well during growth and development.
Epigenetics working on better fix, a gene fix instead of pharmacological fix.
As for time line; note Koch showed Tuberculosis caused by bacteria in 19th century and first antibiotic for tuberculosis mid 20th century.
Note: mTOR fix just stops cells from killing themselves.
There is still very many things that can go wrong. Figure all the things in car that can break and then multiple that number by “n”.
Hi Alan
Did I interpret your website correctly where I believe you said that 40% of CR people are found to have insulin resistance but have low insulin levels and low IGF-1? And those people actually do the best. So therefore the FBS number isn’t all that critical. Did I get that right?
Hi Paul,
The 40% refers to people with 40% caloric restriction. They had had level glucose intolerance as shown by glucose tolerance tests; but they had very low levels of IGF. They resembled male mice with 23% increase lifespan. However, companion group who were long distance runners had no glucose intolerance.
Note that lab mice in these experiments are very inactive compared to mice in wild.
Blagosklonny calls this elevated glucose benign. However, I like to reduce glucose by promoting insulin sensitivity with metformin and walking.
Elevated glucose can cause increase in non enzymatic glycation which can increase risk cataracts. However, low mTOR prevents age related macular degeneration which leading cause blindness in old people. So on balance good for eyes. Nevertheless like to prevent any glucose intolerance.
Thanks
I see what you’re saying. I’m anxious to get my insulin and IGF-1 numbers back
Hi Alan,
Thanks for your explanations. I think I mostly understand and agree with all of what you said.
I will reread Blago 2006 and others of his papers. I think I need also to better understand the whole TOR pathway.
I agree that the time-line for anti-aging treatment based on epigenetic reprogramming could be very far away. I really don’t know. In an optimistic case, it could be 20 years from now though. Do you dismiss that possibility?
Hi Aldebaran,
20 years from now ?
I follow great philosopher Yogi Berra, “very hard to predict what will happen, especially when involves the future.”
Hi Paul,
My current understanding is that it is important for aging to keep level of insulin low but Alan is the expert. This is why I am currently interested in acarbose. Male mice on acarbose live significanlty longer according to the ITP but they don’t have lower blood sugar than control nor they have level of H1b significanlty lower. However, they have insulin level much lower than control.
Hi Aldebaran,
Insulin level number 1 enemy. High insulin 2 huge problems.
First, high insulin causes high mTOR and causes hyper aging as seen in diabetes.
Two, insulin secreted with AMYLIN in 20:1 ratio..Amylin good for glucose control; but high Amylin acts like misfolded protein.
High levels Amylin destroy Beta cells.
Amylin major player in cause type2 diabetes. The amyloid deposits in beta cells in type 2 diabetes is Amylin.
So prevention type 2 diabetes all about low insulin output which means low Amylin output.
Life extension by Rapamycin, caloric restriction of walking perhaps 10 miles a day is all about low insulin and low mTOR.
Thanks Alan for all these informations about insulin and Amylin. This is one more reason for me to walk more. I love walking but just need to find more time for it.
Hi Paul,
There might be a more recent paper from ITP about acarbose but this one is interesting:
“Acarbose, 17-α-estradiol, and nordihydroguaiaretic acid extend mouse lifespan preferentially in males”
In particular, look at figure 3:E (insulin level on males). For me, it was really interesting to see that.
There are on-going life span experiments on acarbose so I expect some updated data soon (likely 2018)
I have to really check into this. Acarbose is more of a European drug, more metformin in the United States. Will look into it . For me insulin, IGF and crp are where the money is.
Hi Aldebaran
Acarbose causes failure break down complex carbohydrates. So life extension looks like results from caloric restriction.
In humans causes malabsorption of carbs.
Also causes severe flatulence and diarrhea.
Here is alternative to a acarbose; eat less carbs.
The idea of eating lots of carbs then taking acarbose so can’t digest seems misguided.
Hi Alan
Big difference mice and people. Give acarbose to mice they get less calories, so caloric restriction.
Humans control their own intake food intake; so people can just eat extra food to make up for malabsorption carbs.
Hi Alan,
Here is a bag of candy, I know I shouldn’t eat that bag of candy.
I just ate the bag of candy.
Ok, now I just take acarbose.
Then just have diarrhea and flatulence.
Still hungry.
Go out for donuts.
Hi Alan,
Thanks.
based on what you said about acarbose I will not consider taking it. Will try to reduce my carbs instead to lower insulin.
I don’t remember the details but I think the ITP guys do not completely agree that it is a CR mimetic. They highlight some differences with CR. I will reread to better understand their point but will stop consider it for myself.
Good to have experts on this site!
Hi Alan,
I understand your point about acarbose. Just want to mention what the ITP said about difference with CR to make sure I am not missing anything.
“Acarbose reduced weight more in females than in males (Fig. (Fig.4).4). Thus, the lengthened survival for ACA-treated males vs. ACA-treated females cannot be explained by changes in body weight or seen simply as the effect of overall caloric restriction. It is not currently known whether slower weight gain is a direct effect of loss of caloric content absorbed or represents a modulation of CNS and gastrointestinal endocrine circuits, perhaps with modification of appetite and associated metabolic set points. The data in Fig. Fig.33 provide an initial indication of key physiological parameters in young adult ACA-treated mice. The lower fasting glucose values, in combination with the unaltered HbA1c levels, are consistent with the idea that ACA may diminish the amplitude of postprandial spikes in plasma glucose levels, with lower peak levels but higher trough levels in both sexes. In both blood levels of FGF21 and activity, effects of ACA differed from effects of DR (Fig 3C,F), and thus benefits of ACA on lifespan may not be attributed simply to diminished caloric intake.”
I thought that it was an interesting study on acarbose Aldebaran. It has a unique mechanism of action in that it inhibits glucose release from complex carbohydrates, I haven’t seen that before. The bottom line seems to be that it reduces post prandial glucose spikes and also lowers insulin. These after meal spikes of glucose and insulin are very dangerous in that they expose the body for several hours to high levels of both substances, so the control of these spikes may be the anti-aging benefit, I don’t really know.
Life extension has a product that combines maqui berry with a clove extract that has been shown in humans to prevent both glucose and insulin after meal spikes to a large degree. Also lowers hba1c levels. May be a similar but safer product.
Hi Paul,
I do not disagree with any of any of your points. I did not study paper. My point is this is not an anti-aging drug. This is drug that interferes with glucose absorption and causes malabsorption syndrome related to dose. It’s apparent effect is lowering glucose spikes, lowering insulin and mTOR spikes and lowering calories absorbed. All these look like good things; but better ways to accomplish those goals.
The study makes a better case for principle to eat low glycemic index foods than to take acarbose.
Hi Alan,
Also, I am not terribly tempted to experienced the side effects you have mentioned with acarbose. I knew about it but was not sure about the magnitude. Looks like to be a non-starter the way you describe it. Not sure why it is popular in Asia and Europe.
Hi Paul,
My understanding of the study was similar to yours and in particular I was very interested by the modulation of postprandial spikes in plasma glucose. However, since I am not an expert, I tend to be very cautious about my own conclusions, but I am really glad that you find it interesting too.
However, side effects of acarbose do not look particularly attractive so if Life extension has a product with similar mode of action and which does not even require a prescription, then I am definitely interested. Do you happen to remember which product it is?
I agree
It’s called glycemic guard. I’m getting it also since rapamycin has modestly increased my glucose. I’ll combine it with Ceylon cinnamon for a synergy.
Controlling insulin and glucose spikes might be a big deal
Gymnema sylvestre
Isn’t that an AMPK activator that comes as a tea?
There are many studies that show benefit for weightloss, AMPK activation but known for optimizing glucose metabolism. Can be taken as a tea. This side of the world prescribed by Natural medicine practitioners for pre-diabetes 2 and diabetes 2.
Thanks
I have ordered glycemic guard from LE. Interestingly, it is just one pill a day, not one pill before every meal like acarbose. It seems to have same effect as ACA but of course we have no mice study to prove it. Will give it a try.
Akshay, I read you were mentioning Gymnema sylvestre. Is it supposed to be more efficient?
Aldebaran it’s an ancient herb this part of the world my father has it with milk every night – he does not have diabetes whereas his mother (my grandmom) died due to diabetic complications. AMPK is also inversely related to mTOR. It is another option for you to study. If you find the time please read up on its various studies to get better idea of its molecular action and side effects if any.
Alan,
you might be right that eating low card diet is same as taking ACA. However, to my knowledge, low card diet has never increased mice life span (in particular max LS). It could be, as you mention, that ACA acted as a CR mimetic but then it should have increased both male and female LS in similar ways. Also, why insulin levels of females did not drop similarly to males? Maybe an artifact of the experiment? I really don’t know but I find it puzzling.
Hi Aldebaran,
Took a look at data.
All about weight loss.
Example at 18 month males 11% and females 23% lighter than controls.
Males better median survival, both about same maximum survival. Both reduced IGF.
Many early deaths females.
So increased survival due to reduced IGF. IGF called grim reaper so no surprise. This CR as seen weight less than controls.
Much more effect females which killed a lot of females so less median survival males.
Interesting part study was reduced weight caused death lot of females who more sensitive to effects drug.
Just another CR effect plus killed more females than males.
We know that women suffer more complications from diabetes than do men, so perhaps the inhibition of post-prandial glucose spikes from acarbose isn’t potent enough to offer a protective effect for females who are more sensitive to the deleterious effects of glucose. Just a guess.
Hi Akshay,
Just quickly checked on Gymnema Sylvestre on webmd (not sure if it is a reliable site BTW). However, they mention one good thing (decrease absorption of sugar) and one bad thing (might stimulate insulin production). Might be good to fight diabete on short term but not so good for aging. We probably need to double check with other sources as I don’t know how reliable this info is:
“How does it work?
Gymnema contains substances that decrease the absorption of sugar from the intestine. Gymnema may also increase the amount of insulin in the body and increase the growth of cells in the pancreas, which is the place in the body where insulin is made”
Aldebaran my understanding is the action is rejuvenative not harmful. So as we age or as part of harmful effect of metabolic syndrome we lose pancreatic cells and therefore reduction in availability of insulin in response to sugars. Nevertheless it is always better to conduct thorough research before ingesting any herb.
Alan,
Thanks for looking at the study about acarbose!
Akshay,
You might be right: we need to look at it more seriously than what I did.
Great discussion all.
Yes it is strange that acarbose has a life extension affect, given we know cutting carbs generally doesn’t help, but cutting proteins does, hence why rapamycin works but blocking protein synthesis. Incidentally it is really hard to cut protein from your diet, plus I don’t think you’d want to.
I think acarbose is a good way to lose weight if you really needed too. Probably even more effective than Berberine or metformin, albeit with unpleasant side effects. Another possibility is glucosamine, which competes with glucose in mitochondria, and is shown to reduce mortality in HUMANS.
‘Use of Glucosamine and Chondroitin in Relation to Mortality’ 2012, Bell et al.
As Alan says this is all about stopping your cells becoming senescent once they have stopped (or slowed down) cycling. Any measure we can take to reduce MTOR, insulin or IGF will do this.
Of course it would be much better if we could keep those cells cycling at the speed they used to. Then we’d be truly becoming younger, not just fighting aging as it accelerates into old age.
This study really makes me wonder if insulin lowering and inhibition of post-prandial spikes is the common denominator with many of these interventions such as acarbose, metformin, berberine, CR, IF , and even rapamycin which can lower insulin and IGF ( though TOR is of course the major mechanism). Could this be synergistic perhaps with the AMPK effect, or maybe AMPK is a red herring and it’s all about keeping insulin controlled, at least in regards to median lifespan.
Hi Mark,
I agree that only reducing protein (and particularly methionine) extend LS in mice in an isocaloric way. There is a great paper from Gustavo Barja 2017 explaining all of that. I remember also a study from Fontana et al. on human CR volunteers which showed that only those who had a reduced protein intake had lower IGF levels (than non CR people).
My take is that it is probably a good habit to avoid eating an excessive number of proteins. Going beyond that would require a careful control and would be too complicated to implement for me.
I do take glucosamine based on the paper you mentioned and on another one showing LS extension on mice, which I have found convincing enough.
Something is still not completely clear to me about acarbose. The ITP has already completed 2 LS study (the one I posted in 2009 + another one in 2012 where mice are started on ACA at 16 months). And 3 more are on going. They certainly think something is going on. I am not interested anymore in trying ACA but I would like to understand better their motivation.
Hi Aldebaran
There is a fair amount of data now on the life extending benefits of methionine restriction (MR), but it seems controversial whether overall protein restriction is also necessary, with seemingly contradictory findings ( McIsaac ,2016,) ( Lee , 2016) ( Speakman) . Th study by Barja suggests that it’s a similar effect on mitROS as rapamycin.
Orgeron 2014 et al showed that ” MR also reduces adiposity but does so through a paradoxical increase in both energy intake and expenditure” suggesting a separate pathway from CR.
How could you reduce methionine without reducing total protein intake I wonder?
Methione is needed to copy DNA, so you can see why it is so effective at slowing aging.
But it is almost impossible to cut it out of your diet, that is why rapamycin is such a good option in comparison because you don’t get the horrible cravings you’d get if you actually did manage to cut it down. Plus is would be very hard to do it intermittently. Better just to intermittently fast and get used to that, I think.
I am not sure if any other these paths are independent. MTOR and AMPK are just opposite sides of the same coin, only MTOR is much stronger. Hence all the AMPK activators are inferior to rapamycin, as far as I can tell. It would be really nice to find a completely different pathway. Might have to look back into those worm studies for that.
Hi Mark,
Totally agree very exciting to find pathways extend lifespan outside mTOR, insulin pathway.
Angiotensin 2 blockers seems to be separate pathway involving mitochondria.
AMPK seems to partially separate action mitochondria.
Also very many indirect inhibitors mTOR.
Mark,
Paul and I have looked recently at the following studies in worms:
“Slowing ageing using drug synergy in C. elegans”
They had pretty interesting LS numbers using combinations of Rapamycin + Allantoin + Rifampicin on worms and flies
It seems that the mode of action of Rifampicin is completely different than Rapa. In the following study they claim that RIF extends life span of C.Elegans through AGE inhibition:
“Rifampicin reduces advanced glycation end products and activates DAF-16 to increase lifespan in Caenorhabditis elegans”
However, this is on worms/flies so probably need to take that with a grain of salt.
Hi Alan,
ARBs look indeed very interesting for anti-aging. Are there any side effects? In particular, can they lower the BP of people with already low BP?
Thanks
Hi Aldebaran,
Hypotension limiting factor. Can’t take if normal blood pressure due to risk hypotension
As far as i am concerned MTOR inhibition (either direct or through AMPK activation), and mtROS are independent drivers of aging, although many pathways such as MTOR and AMPK appear to affect both somewhat. It would be interesting to see if angiotensin inhibitors/blockers use a different pathway to lower ROS. There are effectively two ways to do this. Either lower endogenous ROS production (best), enhancing mitophagy does this for example, or increasing antioxidant defence, either exogenously, throuh say a mitochondrial antioxidant, or endogenously through a supplement that activates Nrf-2.
I don’t take any angiotensin inhibitor because I think the mitophagy I do through Nicotinamide and Ribose serves the same purpose, but I could be wrong.
I also have the idea that increasing adiponectin could be a real winner for several pathways, though I’ve yet to try that yet.
Unfortunately I wouldn’t expect an AGE inhibitor or breaker that works in worms and flies to help humans, as we live so much longer we build up crosslinks they never have time to experience. Don’t forget the drug Algebrium worked in everything up to and including monkeys, but nada with humans.
Hi Alan,
Thanks for pointing out limiting factor about ARBs and BP. Will definitely consider it if my BP raise.
Mark,
Good point about AGE breakers. I am not sure if the mode of action of RIF and Algebrium are similar though (Algebrium is it ALT 711 right?) :
“For its effect on longevity, rifampicin requires DAF-18 (nematode PTEN) as well as JNK-1 and activates DAF-16, the FOXO homolog. Interestingly, the drug treatment modulates transcription of a different subset of DAF-16 target genes, those not controlled by the conserved Insulin-IGF-1-like signaling pathway.”
I would still be interested to see what RIF does on mice.
Hi Paul, yes you are definitely on to something. At the cellular level a senescent cell is basically an old cell that can no longer divide. It doesn’t take too many of these to kill an organism, so anything that slows down the accumulation of senescent cells has to be a good thing, no matter the pathway. It remains to be seen how effective senolytics is, as it’s success will largely depend on how well stem cells can be mobilised to replace the removed cells. That is why extending telomeres is preferable as it also acheive this aim.
Paul Rivas,
On the other hand there are many other plants, besides ashwagandha (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4658772/), presenting in studies positive effects on the autonomic nervous system and/or in reduction in lactic acid production. Among these plants are quercetin, resveratrol, curcumin and crataegus oxyacantha (Hawthorn).
Hi Mark
I think you’re certainly correct.
In terms of maintaining cells with telomerase activation it would seem that we’re close to achieving that end.
Adding senolytics to rapamycin might be a good synergy if it’s not overkill.
As for stem cell therapy, we may be getting closer there as well.
You may recall that they injected MSC’s from healthy mice into mice with osteoporosis, and after 6 months the osteoporotic bone had given way to healthy , functional bone. Stem Cells Translational Medicine . William Stanford 2016
You’ll find this interesting. There is a doctor in California with whom I correspond from time to time. He’s an MD and masters in public health from Columbia Univ., and has written 2 books on telomeres as well as treating over 1000 patients with telomerase activators. He cites evidence that centenarians have a very homogenous distribution of telomere lengths, very few very long or very short, but rather a smooth distribution. Mech Ageing Dev 2008 Nov 129 (11) 638-41.
There is a patient of his named Karl, 72 y/o , with 7000 base pairs. He gave him 4 TA 65’s a day.
After 1 year his median length dropped to 5900 with 25% very shorts
At 2 years he was down to 4900 with 27% very shorts
At 3 years he was back to 7550
At 4 years he was at 11,600 with only 3 % critically shorts.
He is now 77 and has stabilized at that number, apparently feels great and is still working.
Ed explained his initial 2 year drop as this:” If you induce apoptosis in older damaged stem cells, then you get a representation by younger or replacement stem cells with telomere lengths more closely distributed around the original middle of 15kbp for every telomere. With apoptosis and replacement, you don’t have those old and overgrown telomeres around as much. This kind of drop in median length is not uncommon and something that no one else is discussing yet. “
That is interesting Paul, and is consistent with what the recent Ta-65 trial found – a small but significant effect with 1 Ta-65 pill a day (for a year), but no improvement with 4 pills a day. I think that this is probably not a bell shaped response curve, but something else. I’m not so sure about Ed Park’s explanation. I would say it is probably more to do with rescuing senescent cells, with initially very short telomere lengths.
The study: A Natural Product Telomerase Activator Lengthens Telomeres in Humans: A Randomized, Double Blind, and Placebo Controlled Study. Rejuvenation Research , Dec 2016.
I’d say that you are right
He now uses ashwaghanda, rhodiola, holy basil, and curcumin along with ta65 to get more of a bang out of telomerase activation.
Check this out Paul,
‘Identification of Telomerase-activating Blends From Naturally Occurring Compounds.’ Altern Ther Health Med. June 2016.
The blend they settled on was broccoli extract, astralagus, rhodiola and Vit D, and it was comparable or better than Ta-65 at telomerase activation in vitro, in the cell type examined. The components are also far more bioavailable.
Thanks Mark I’ll look at it. With me at least, rhodiola has an initial calming effect, but then seems to accumulate and makes me anxious. Ashwagandha does the same thing.
I’ll have to try it again; already take the others.
So we now know of several substances , which taken together , probably activate telomerase more than ta 65 does,, but we have actual human data showing a telomere effect with ta 65 that we don’t have with the others. What to do ?
I asked Bill Andrews if they had thought about combinations of cycloastragenol with other things to exploit different mechanisms of action of telomerase activation. He said they did and it was called their Synergy project. So even he thinks a combination will yield greater results.
Interestingly their new telomerase activator, TAM818 I think it’s called, from the patents looks to be an altered version of cycloastragenol with much improved bioavailability.
Good information
In my lowly opinion Bill Andrews is the foremost expert in the country on telomerase/telomere science.
Do you recall that he was the first to try TA 65 and he said that the next day he looked in the mirror and expected to see a 20 year old looking back? LOL. Maybe the expectations were set a little high.
I didn’t know Bill Andrews said that, but I’ve heard him say that he’ll not consider aging solved until you get someone like a long faded movie star come back on stage looking 25. In my opinion that is what a proper solution to aging will look like, and Bill is right to be ambitious.
Hi Mark,
This study is interesting. The TL elongation obtained with low dose TA65 looks convincing although the lack of effect with the larger dose is puzzling. This is actually the sort of compound that might work better in human than mice since mice have much longer telomeres.
However, we don’t have yet convincing data showing a positive impact of telomere elongation on human life span do we? Do we even know if replicative senescence is a major contributor of the senescent cells in our body? If not, then elongating telomeres might not be that useful (outside of maybe reducing cancer rates by reducing the proportion of critically short telomeres).
I really like the research around telomere elongation but it is hard to quantify its impact without any life span study.
Well it helped mice and they don’t suffer from replicative senescence at all, so my bet is that it will be very helpful for humans. As Paul has been saying, anything that helps with reducing or correcting senescence is going to help, regardless of the path it took to reach senescence. There are also other things that make me think it will useful, namely a Shay and Wright study showing how gene expression changes from young to old to senescent cells and how telomere lengthening restores almost all of it. Also another paper showing how the majority of the changes in the aging brain are down to glial cells rather than neurons, which matches what Michael Fossel has been saying all along about Alzheimer’s. I’ll try and dig out the papers.
Also there is a mouse lifespan study that acheived quite a significant lifespan extension. Done by Blasco et al, but with a telomerase gene therapy, not telomerase activators, which appear from what I can tell to have very bad bioavailability.
‘Telomerase gene therapy in adult and old mice delays aging and increases longevity without increasing cancer’.
These are good points. I am not confident enough to consider taking telomerase activators yet but definitely something to follow-up.
At extreme ages we definitely suffer from replicative senescence. Just look at old skin or blood. My 98-year-old grandfather-in-law needed constant transfusions because he had stopped making his own RBCs (of course the transfusions are probably was parabiosed him to 98 😉
Confirmation bias warning, I worked for Shay-Wright for five years.
Hi Bill,
But do we know for a fact that most of the cells that become senescent with age do so because their telomeres become critically short?
I don’t doubt that cells become senescent with age but I am just not sure if it is mostly related to telomere length or not.
Hi Bill,
Good joke about parabiosis, a variation of new blood in old bottles. Vampire myths were already old wives tales in 19th century and appear to go back a few thousand years. Naturally it should be the next big thing in anti-aging crackpot medicine.
As regards unexplained anemia in 98 year old; my experience is stem cells making blood, skin, mucosa, liver continue to function very nicely in old age. So If he died from unexplained anemia and I was the Medical Examiner on the case: I would be more likely to order full toxicology to rule out intentional or accidental poisoning, than to write on death certificate, “anemia due to short telomeres”.
aldebaran, the cultured BJs that senesced in my dishes in the lab had shorter telomeres than lower-PD cells. But I’ll be the first to say that you could keep them from senescing for another 20 PDs if you grew them in fetal serum…
Alan, parabiosis (actual parabiosis, not just adding plasma) has been extending the lives of rats since the 1880s. Not to mention Bogdanov’s little experiment (no doubt with blood from the “kulaks”).
Parabiosis works for sure in rodents, and if you cloned yourself it would be hard to understand why it wouldn’t work in humans. (Simply adding plasma may fix fewer problems than connecting a young immune system, liver etc. of course).
Blood cell counts routinely fall in the very old. I doubt that they’re ALL being poisoned for their estates… or that if they were, the poisoners would all be using blood poisons instead of neutotoxins.
Hi Bill,
Are you aware of any study showing life extension in mice with parabiosis? The only one that I have looked at is a life span study on mice receiving plasma transfusion of young blood during all their lives and they did not have longer life than control. My knowledge about parabiosis is close to zero so I am not saying anything negative. I am just interested to look at the facts.
Hi Aldebaran,
Most important point about parabiosis is totally wide open. Blood is legal product, so anybody with medical license can prescribe blood to anybody that will hold out their arm. Any doctor can set up facility to take blood and to administer blood. That means can pay a young person $20 for unit of blood and then give that unit to somebody else and charge them $20,000. Physicians have been doing millions of blood transfusions for a hundred years and nobody came up with parabiosis scheme until anti-aging became the next big thing as regards con games.
As regards research, everything you read in the bible ain’t necessarily so and that goes double for a lot of scientific research.
Color me skeptical.
This study shows that biological aging can be measured accurately by the proteins in the blood.
‘Protein profiling reveals consequences of lifestyle choices on predicted biological aging’.
Shows the benefits of coffee drinking and oily fish, as well as the harmful effects of smoking, sugary drinks/sweets and obesity.
Obviously not as simple as giving an old person young blood. This is more about the state of our blood being a reflection of the state of our body. Hence hooking rats together helps the old rat but harms the young one.
Hi Mark,
Very interesting paper about protein profiling.
I will try to find the time to read it carefully today.
Thanks,
Hi Alderbaran,
re replicative senescene as cause of aging:
I think telomerase might be misleadingly named, Maybe it should be called “immortalase”. It does much more than just elongating telomeres.
Replicative senescence by telomere shortening is just an extra protecion against cancer in long living animals.
But telomerase – immortalase is something that we inhibited from our pre multicellular or pre triploblastic origins. I think telomerase is just a protection against runaway asexual reproduction which is bad for the long term health of the gene pool for the unicellular eucaryotic organism.
Multicellular, and especially triploblastic organisms have another layer of control on top of the telomerase and this layer is most probably epigenetic.
This epigentic layer can control telomerase and causes age related organismal senescent via the p16 pathway. Also the cell loses its multicellular function if it loses its epigentic information through epigenetic aging, which is just a continuation of the developmental program.
So I think telomerase immortalized mulitcellular cells sooner or later lose their multicellular nature because they can no longer maintain their epigenetic state.
Even if they are immortal unicellular beings I doubt they can form functional multicellular tissue ever again.
Cellular aging is probably telomerase (much more than telomere length!) dependent, whereas organismal aging is epigentic in nature.
Hi Gabor,
Thanks for your comments. If I understand correctly your point you are saying in particular that:
1) Aging of multi-cellular organism is essentially controlled by epigenetic changes
2) Cellular aging can be fully controlled by telomerase. However, systematic activation of telomerase cannot be used blindly to rejuvenate multi-cellular organisms because it would erase the epigenetic information encoded into differentiated cells (and so they would end-up losing their identity).
Certainly it seems very plausible to me that aging of multi-cellular organism could be controlled by epigenetic changes to some degree but I tend to think that mitoROS plays also a role in aging. However, it could be that mitoROS are also controlled by epigenetic changes or it could be the other way around. In any case, I find your opinion interesting
Since you mentioned that telomerase control cell aging, is it systematically expressed when cells are reprogrammed (into iPS cells)?
Hi Aldebaran,
I’m just not smart enough to come up with my own theory of aging; so I just follow Blagosklonny and apply his ideas to clinical medicine. Aging appears to be most complex disease in all of medicine; so way too complex for me.
yes, I am trying to pull together the two facts that telomere dependent replicative senscent does not make mouse older, so there must be something epigentic controlling it.
however my claim that telomerase immortalized cells lose their function might not be correct, as there are papers on engrafting telomerase immortalized cell lines into living organisms with success.
unfortunately I have only found one paper that separates epigenetic aging from replicative aging, stating that epigenetic aging continues
“Epigenetic clock analyses of cellular senescence and ageing”
stating that cells continue to age epigenetically after telomerase immortalization.
I contacted the author and he told me that the telomerase immortalized cells
“they age morphologically and epigenetically as they are cultured. “
Very interesting GaborB, but can you explain what you mean by ‘This epigentic layer …causes age related organismal senescent via the p16 pathway’? How is this different to the p21 pathway triggered by replicative senescence?
Also to my knowledge telomerase immortalized cells passaged in vitro have not ever been seen to lose their differentiation; although I’ll admit that is plausible (after all it happens when you go the other way generating iPSCs) it might not happen in a timescale to be meaningful in human aging.
Btw I totally agree with how you’ve explained telomerase as a defence against genetic uniformity in single celled colonies now being used as a defence against cancer in multicelled life. Very well put.
Alderbaran, it is my belief that MTOR and mtROS are driving epigenetic changes both through telomere erosion and more general methylation changes unrelated to the cells ability to proliferate (that is controlled by telomerase). This is how the aging rate is set for different species. The big unanswered question in my mind is how important the methylation changes are in human aging (outside the problems related to shortening telomeres).
sorry, I wanted to reconcialiate the two facts that mouse does not have telomere dependent replicative senescence and in humans epigenetic aging is more correlated with chronological age than telomere length.
I think that in mice MTOR is set so high (grow up and breed quick before you are killed!) that most of their cells senesce simply through replicative stress, regardless of telomere length. MtROS is also really high too so this probably contributes to general stress on their DNA causing senescence due to DDR.
It would be very interesting to see if mice have the same epigenetic methylation pattern changes as humans, but over a shorter timescale. I believe the answer is yes, see Fig 3b from:
‘Caloric restriction delays age-related methylation
drift’
The big question for me is are these epigenetic methylation changes just a clock, or are they a cause of aging?
It may be cause for aging as these methylation changes seem to be silencing promoters of beneficial genes and unlocking the damaging ones – someone here had mentioned about Transposon Elements being unleashed. It seems reversing or resetting the epigenetic change can instantly show the benefits of expressing a helpful gene. As an example I would like to cite a 2016 study by Dr. Jean-Pierre Issa of Temple University Targeting Calcium Signaling To Induce Epigenetic Reactivation of Tumor Suppressor Genes in Cancer. The reactivation of the TSGs through reversal of the epigenetic silencing has shown action against colon, glioblastoma cancers in multiple studies.
Hi Mark,
p21 and p16 pathways are somewhat independent as described in this paper
“Mechanisms of cellular senescence in human and mouse cells”
I share your doubts about telomerase immortalized cells. I yet have to find concise evidence whether they have runaway epigenetic aging and whether it matters. I have only found one paper yet that investigates both epigenetic aging and telomere dependent senscence.
It might well be that these other (non telomere inhibited) epigenetic changes increase the change of cancer, I posted a paper in the discussion downstream somewhere about ROS causes epigenetic downregulation of catalase expression then allowing metastasis via the mitochondria.
You wouldn’t see this in vitro (too few cells), but it would become relevant in vivo.
Just speculating at this point.
There has been a great debate in the 2000s over what is required for cell immortalization. Is telomerase alone sufficient, or is p16 inhibition also necessary.
I think those experiments should be repeated in light of the findings of the epigenetic clock.
My current hunch is that epigenetic clock should be lethal in itself because of epigenetic de inhibition of transposable elements. But if it is not the case a second guess is that epigenetic clock is sensed by p16. The third guess is the epigenetic clock simply measures the loss of stem cell plasticity that determines the functional fitness of the daughter cells in tissue and p16 measures this.
According to the paper you posted on p21 and p16, some cells are extra sensitive and sometimes senesce early before getting short telomeres. They don’t know what the mechanism for that is in that paper, but from reading around it seems p16 is mainly used by cells to detect oncogene activation, as opposed to p53/p21, which is a more general arrest on cells (often but not always due to replicative senescence). Suppressing p16 in vitro allowed such cells to continue on to final p21 mediated replicative arrest.
I think your idea has merit. If it is true then in those sensitive cells that more often produce p16, even when immortalized by telomerase, should gradually lose proliferating cells to p16 arrest. Blocking p16 would prevent this loss, but perhaps at the cost of cancerous transformation in cells (eventually). This fits in with what both Akshay and I have said about epigenetic changes being relevant to cancer.
The question still remains about how much this matters, i.e. if all the cells in our bodies were periodically refreshed by telomerase, would we still age due to other arrest signals related to non telomeric epigenetic changes.
Wow.
I went looking and immediately found a paper that completely supports your idea (at least in those sensitive, p16 prone WI-38 cells).
See ‘Prolonged Culture of Telomerase-Immortalized Human Fibroblasts Leads to a Premalignant Phenotype’.
Note the part where they show telomerase immortalised WI-38 cells have much lower colony forming efficiency then before immortalization, suggesting something else is stressing them (causing a p16 response perhaps?), until about 250 passages when the cells that were successful start to dominate and look more and more like cancerous cells.
This to me looks like a tentative validation of the idea that even after telomerase immortalisation, cells eventually face the choice of senescence (aging) or cancerous transformation.
Yes I have read that article before but there is also a rebuttal.
“Absence of cancer-associated changes in human fibroblasts immortalized with telomerase”
I think a lot depends on culture conditions and cell types whether there will be a malignant transformation or not.
But in general I believe p16 has to be overcome for telomerase immortalization. In some cultures the researchers silence p16, in some cultures hTERT expression makes p16 methylated somehow.
“Methylation of the p16INK4a promoter region in telomerase immortalized human keratinocytes co-cultured with feeder cells”
In some cultures p16 inhibitor bmi-1 is overexpressed.
I have found a study where they had an intact p16 dependent senescent pathway in telomerase immortalized culture, yet cells went into senescent when they forcibly expressed p16.
“Telomerase Induces Immortalization of Human Esophageal Keratinocytes Without p16 INK4a Inactivation”
So I think they disrupted the pathway acting against immortalization in another way or maybe the epigenetic clock was not advancing.
These are all decade old articles I believe without a concise conclusion. I think there are a lot of low hanging fruits there that could be reaped using the epigenetic clock with relatively low research budget. Although a whole epigeneome investigation alone costs some 10k dollars. + the cell culture research. but still much cheaper than mouse models imho
Yes, maybe you are right and this is just an artifact of the cell culture and cell type being ‘trained’ to go down a certain developmental path, i.e. do not heed contact inhibition.
We need these experiments repeated with multiple cell types by Horvath’s team to see what is going on at the epigenetic level. As far as I am aware he has done this only up to 50 passages of endothelial cells and observed that when immortalized they did not senesce but that they continued to age epigenetically. Perhaps it is not possible in vitro to determine what would eventually stop them for the reasons we’ve discussed above.
My current understanding is that protein synthesis increases with age because ribosome biogenesis increases with age due to epigenetic changes (in particular histone modifications). This is one reason why I think TOR should be inhibited with age (to counterbalance the increase in protein synthesis). In that sense, epigenetic modifications are exacerbating TOR.
Therefore, if we were able to reverse epigenetic changes, we would likely restore TOR signaling to a normal level. In my view, it would be a better option than to inhibit TOR signaling.
This is a nice article that support and expands on the one we discussed above:
‘Immortality, but not oncogenic transformation,
of primary human cells leads to epigenetic
reprogramming of DNA methylation and
gene expression’.
The title is misleading. It is basically saying that immortalized cells gain methylation over time and that some of it favours their long term survival and plasticity. The most interesting point however, is that most methylation occurs on sites that are already inactive in the non-immortal parent cell line, as is essentially stochastic (as I have long argued).
So I am back where I started. The Horvath clock is looking more than ever like a selection of epigenetic changes (certainly the hypermethylated part) that are not important to a cell’s survival. Otherwise they would not be permitted. The remainder are probably cell adaptations to conditions rather than drivers of aging. How does the Horvath clock independently predict mortality then? Probably just through an association or correlation with other aging factors.
Most cell culture is done under completely crazy non-physiological conditions. Cells in the body are at 1-2% O2, human instead of cow serum, attached to other cells not plastic….
there’s a limit to what you can conclude from watching cells in culture. But I will say that telomerized cells grow a little faster than the untelomerized, in many, many different species.
Hi Bill,
why people dont try to understand what lies beneath the differential behavior that depends on the cell culture. as an outsider I would think a lot of knowledge could be gained this way.
Also do you happen to know why nobody does iPSC generation with C. elegans? I found zero articles about this.
For those of you who haven’t seen it yet, this is a very interesting talk by Steve Horvath about the epigenetic clock from last year:
https://youtu.be/0zaCKAnFogQ
Some quick take-aways:
For Horvath, this is more of a developmental clock than a biological age one. But it can fit both descriptions well.
Cancerous cells tend to exhibit much older profiles than normal cells
There doesn’t seem to be a correlation between average telomere length and epigenetic age.
Progeric children do not seem to age faster according to this measure.
Men tend to be older than women (due to large body size perhaps?, this is my idea, not Horvath’s) for the same chronological age. It would fit with the average shorter lifespans of most men.
Breast tissue in women tends to be significantly older than others. Is there any relation to the prevalence of breast cancer?
There is a correlation with a high carb diet and epigenetic age, but acc. to Horvath it is weak.
Right after differentiation iPSC’s already show measurable age epigenetically.
Thank you Adrian for sharing the highlights – I am a fan of Horvath. I am wondering if he has seen any change post puberty. As per recent studies many aging dysfunctions seem to starting just after reproductive organs are formed.
Horvath is a clever guy, and his method or those derived from it are undoubtedly a very accurate measure of age across tissues. But, and this a big one, there is currently no link betwen this measure of aging and the diseases of aging. It could well be that there is not any causation here at all, I.e. methylation just happens in a fairly random way, so it increases with time, but this is coincidental with aging. Or it could be that it causes some problems, but that these are not a big issue to the ages we currently live to. For example telomerase immortalised cells continue to clock up epigenetic decorations but this doesn’t impede them from continuing to proliferate in any way. So this is probably why cancerous cells appear older by this measure, not because they have become cancerous because of this epigenetic age necessarily.
On the other hand we know the rate of epigenetic drift is set by MTOR, as it can be slowed by slowing MTOR, so perhaps a link can be established to premature senescence and that would go a long way to making Horvath’s work really useful other than just as a measure.
We do not have anything close to a cure for aging. Inhibition of mTOR improves healthspan and hopefully some incremental lifespan will come to know when the pioneers to start rapamycin cross 100. In the meanwhile all of us have our pet theories. I am not negating any other theory because it could be right. But wanted to share my thoughts. I know you currently seem inclined towards telomere shortening as a cause for aging. I am currently inclined towards looking upstream for the cause and so epigenetic migration from puberty onwards seems where all dysfunctions downstream appear from. Of course what triggers the aging related epigenetic changes is yet unknown to me. Any of the dysfunctions when alleviated do show benefit and so seem to be a cause instead of an effect. I want to cite a recent study by Dr. Gary Samuelson called ‘All in the Genes – Redox Signalling and another one by Thomas Wilhelm ‘Neuronal Inhibition of the Autophagy Nucleation Complex Extends Life Span in Post Reproductive C.Elegans’. We have always considered autophagy as a beneficial process that dials down with aging and is a cause for accumulation of senescent cells which in turn have been shown in earlier cited study today as the starting point of all aging dysfunctions. But in Thomas’s study they found that by inhibiting autophagy in old age there was significant improvement in lifespan and healthspan. The reason given for this paradox is that as we grow older the dysfunctions in our beneficial processes grow to such an extent the same become a major cause of damage to an extent where inhibiting them revives health and extends life. So the various effects or symptoms of Aging when acted upon would mitigate the damage caused by it and since everything so interconnected it has almost systemic benefits. Yet we can only expect booster dose from such interventions which too are most welcome but hopefully one of these days someone will find a way to reverse or stop the aging process at the very source whatever that may be. Till then every safe intervention that shows health benefits in human clinical trials are what may keep us alive till when the ultimate cure arrives.
I agree with you Akshay, I think we are still not seeing the big picture but – perhaps we are not too far away from the time when these diverse theories start to come together.
Personally at the moment I am of the opinion that Blagosklonny is closest in that the faster we are programmed to develop the faster all our biological systems seem to drift out of calibration as we get older. Whether this is a stochastic process with ROS damage to the everyday proteins we produce to live, or whether it is due to some sort of epigenetic deregulation of repair processes after puberty, or both, I just don’t know. There are clearly some evolutuonary constraints though, like the SNPs for longer telomeres being dangerous for cancer and us needing senescent cells for wound healing and as a barrier to cancer. And these constraints need not apply to medical interventions, so we are already on the cusp of being able to reset these parts of aging, whatever fraction that might be.
Judith Campisi was interviewed in a prior issue of life extension magazine regarding senolytic agents and she feels that clearing senescent cells with these agents once every 5 years would be optimal. She claims that senescent cells develop slowly enough to allow for that time frame.
Yesterday I took a brief look at the Horvath clock.
Two notes:
it uses Illumina27k data, so mostly promoters. I think no transposable elements are included in the raw dataset. So it cannot capture the well documented age related demethylation of transposable elements.
Looking at only one sample even thhough the data is not normalized, it looks like both the methylation and demethylation sites change the most at young age. So I believe Horvaths clock captures the epigenetic development program and he got lucky because this program continues into adulthood although in a much slower pace.
I wish I had some time to play around with the raw data, I think Illumina450k or Illumina850k might have a bunch of transposable element and telomeric sites.
Hi GaborB, question about epigenetic clock:
According to Blagosklonny aging theory, growth and development is very tightly programmed; therefore growth and development would need a kind of biologic clock.
However, according to same theory, aging is not programmed, so aging would not need a clock.
According to antagonistic pleiotrophy theory, once epigenetic clock was set up for growth and development; in older persons it would continue to run even if it had no purpose. Furthermore, it could cause harmful effects.
Do you find anything to support this antagonistic pleiotrophy type hypothesis; that in growth and development biologic clock is essential; but in aging biologic clock is detrimental.
Hi Alan
I think Horvaths clock is a strong clue for pleiotropy. But really I dont know what those promoters do and in which tissue.
Actually I think aging is programmed in a passive way. The organism could have evolved ways to overcome pleiotropy, but letting the developmental program run its course and become detrimental to the organism was evolutionary more favourable.
Hi Mark, you are right it’s too early to tell whether this is just correlation. But I wouldn’t say this is a random process. Quite the contrary, or the whole concept of an epigenetic clock would not be possible. It may well be ‘drift’ driven by the mTOR pathway after puberty, but it still seems to be very predictable. The only way I can see this being random is if the methylation patterns are caused by chromatin areas which are more exposed than others. But this doesn’t seem too likely to me. Among other things, the pattern is reset in iPSC, so the cell seems to be in control of this process, at least to turn back the ‘clock’.
I have to say I am a bit bummed that there is no correlation with average telomere length. Perhaps this is an example of the multiple ageing clocks Josh has proposed many times.
We need to get more granularity on exactly what is happening at different times in differing tissues with epigenetic changes. Exactly how much of this is important in terms of differentiation say, and how much is what I term random drift on those CpG islands. A process can definitely be random but still totally predictable, that is how atomic clocks work.
Horvath’s theory is that the epigenetic changes are related to the work done by the cell to maintain epigenetic expression, which would explain how it happens faster in faster proliferating tissues, but also happens in post mitotic cells.
I agree with Mark that there exist random processes with overall perfectly predictable outcome at the global scale. For example radioactivity: the decay of a beta particle is a random process with fixed probability but if you have enough particles, you can predict with high accuracy when half of the particles will have decayed.
In the cases of the 353 CpGs defined by Horvath, it is more complicated because it contain 193 CpG that become hyper-methylated with age and 160 that become hypo-methylated, so you’ll have to assume that the probability of CpG to become methylated or unmethylated is non uniform.
Even if the methylation / unmethylation of the CpG happen randomly, it can still be a causative factor of aging. For example, if CpG islands inside promoter regions of Transposable Element become hypo-methylated with age, these TE will be expressed which could disturb the cell function and increase its mutation load.
I actually would not be surprised if DNA methylation has a causative role in aging simply because it could promote the expression over time of initially silent TE. However, to my knowledge, Horvath has not investigated that point.
That’s a very good point Aldebaran. Are there any studies of the role of TEs unleashed in aging?
Hi Akshay,
Yes, there several recent publications about the role of Transposable Element in aging. In particular this one:
“Chromatin-modifying genetic interventions suppress age-associated transposable element activation and extend life span in Drosophila”
Thanks!
This study may be of interest:
‘Downregulation of catalase by reactive oxygen species via hypermethylation of CpG island II on the catalase promoter’ edited by our old friend Vladimir Skulachev.
In this paper they demonstrate that in a particular cancer hepatocellular carcinoma (HCC) prolonged exposure to ROS causes hypermethylation of the catalase promoter region, allowing further increases in ROS and metastasis of the cancer.
We already knew cancer had to hijack mitochondria to spread, and although the mechanism is not yet clear we now have reason to believe it is via epigenetic methylation.
More and more I cannot escape the notion that a species rate of aging is set by MTOR and mtROS and everything else is downstream of these fundamentals. Of course reducing MTOR and mtROS can only delay/slow down aging. If we want to reverse it we must look to reset the changes in telomeres, epigenetics, TEs, etc, that MTOR and mtROS have wrought.
Hi Mark,
Interesting link between ROS and DNA methylation. The caveat is that they have only looked at one CpG Island (catalase promoter) and only in-vitro (and only in HCC cells). It remains to be seen if ROS (particularly mitoROS) is a systematic cause of DNA methylation / demethylation. If yes, this could open new possibilities to slow down the epigenetic clock.
Yes you are right, these findings are very specific and only preliminary. However the weight of evidence in general is that the aging rate of species is set by MTOR and mtROS (mtDNA stability) as shown in the following paper:
‘Do Mitochondrial DNA and Metabolic Rate Complement Each Other in Determination of the Mammalian Maximum Longevity?’ Rejuvenation Research, 2008.
And we know epigenetic methylation patterns track aging extremely well, so it is not a huge leap to come to the conclusion that MTOR and mtROS are causing the epigenetic methylation patterns to change.
In my mind the question is: do these epigenetic changes then lead to aging, and if so can resetting them reset aging? Early signs from the work done by Belmonte et al, and reported in Josh’s blog, tentatively suggest the answer may be yes.
ROS driving DNA demethylation absolutely makes sense. There are a lot of papers on the 3 or 4 step process of the oxidation of methylcytosine back into normal cytosine.
The other part of the epigenetic clock is more tricky IMHO. That part I believe is probably governed by changes in histone modifications.
Basically I believe histone modifications might expose DNA parts to methylation enzymes and the stochastic demethylation process as well.
Actually I am really curious wether the two parts of the loglinear Horvath model are really loglinear the same way. It may be that the two processes are completely separated, there is a development process with stepwise metylation of the developmental genes dominating early life and there is a stochastic demethylating the whole genome in an oxidation dependent manner.
It might also happen that the methylcytosine oxidation component is the one giving cadence to the development program.
Hi Gabor,
Do we know the cause of histone modifications? Could it be ROS and particularly mitoROS?
Also you said that histone modification can cause DNA methylation / demethylation but could it be the other way around? (could DNA methylation /demethylation be the cause of histone modifications?).
It is weird that some sites get methylated and others get demethylated. You would expect either methylation or demethylation to predominate.
Perhaps the histone modifications are driven by down regulation of deacetylase inhibitors like Sirtuins, opening up more of the genome to modification. This would provide a nice link back to mitochondria and ROS.
I sure this fascinating area will keep be reading papers for some time to come…
Yes I agree this looks weird. But as long as these changes contribute to increases the entropy of the cell, this is perfectly expected.
For example, if the lowest entropy is obtained when the ratio of methylated G is equal to RG, you would expect that every CpG island more methylated than RG will get less methylated with time and every CpG island less methylated than RG will get more
methylated with time.
I am sure I am oversimplifying the picture but this is just to give an example of how it could go both ways while still increasing the entropy in both cases.
I dont have any solid wisdom on histone modifications. I have collected a bunch of articles on my aging blog.
This one is outstanding
“Epigenetic regulation of ageing:
linking environmental inputs to
genomic stability”
What is your blog Gabor?
Unfortunately it is not open access. I have found this one instead, which is open access and looks informative:
“Epigenetics and aging” Sci Adv. 2016 Jul; 2(7)
Gabor do you like this one too?
Yes that is a plausible explanation for why you might get some sites methylated and some sites demethlyated over time Aldebaran. I do not think that is what is happening however. I suspect that methylation is largely random, just tending to increase over time, but in general not having any great effect on gene expression. Demethylaton I suspect is mostly programmed, with cells keeping clear what they need to promote and maintain the required gene expresion, but with some stochastic demethylation too based on things like ROS.
I’m not an expert at all on epigenetics, but I’m getting this idea from lots of different papers.
Mark,
You know more than me on epigenetic and your explanation make sense. It would explain why we see both methylation and demethylation at the same time. I am currently reading some papers to get a better idea of what is going on. It is really a fascinating area and I suspect that our understanding of epigenetic and its implications in aging is going to dramatically improve in the years to come.
Extract from the paper above:
” For example, heterochromatin decay and histone loss lead to retrotransposition and changes in gene expression. But what causes the heterochromatin decay and block in histone protein synthesis during aging in the first place? We are currently limited in our knowledge of the sequence of the causal events during aging in healthy individuals. Attempts should be made in the near future to define the cascade of events during age progression to attain a comprehensive view of the aging process and to identify the initial causative events”
Extract from the paper above:
“Epigenetic drift leads to unpredictable differences in the methylome among aging individuals (Fig. 3). However, some of the methylation changes that occur with age are directional and involve specific regions of the genome. This fact indicates that at least part of the DNA methylation changes during aging are not stochastic but could be associated with biological mechanisms involved in the aging process. “
Hi Aldebaran,
you can access my blog if you clock on my nickname.
understandaging.blogspot.com
My understanding is that the epigenetic clock of Horvath is not stochastic. There is genuine demethylation and methylation going on as aging progresses. I think these are governed by histone modifications.
There is a stochastic demethylation as age progresses and it affects TE sequences. But Horvath’s clock does not address those.
If you cannot access an article, try scholar.google.com and search. Under each article there is a link “All x versions”. Clicking on that might reveal links to free copies. And then there is researchgate.net where you can ask a copy from the author. usually they give it away. and then there are russian and chinese webpages where you can find anything
Hi GaborB,
I find your blog interesting and informative. I will spend more time on it and will also read the attached articles. Also, thanks for the tips to get the articles.
It is just so frustrated how little we know about these epigenetic changes. I’ve changed my mind numerous times on the importance of the sites included in Horvath’s clock. But based on the papers I’ve read where they immortalized cell lines and passaged them for a really long time, it appears to me that the locations he has included are so ubiquitous across tissue types precisely because they are not that important to function. I am happy to be proven wrong, let’s wait and see what Horvath is working on.
Hi Akshay. He more or less addresses that question. At about minute 15 in that video he shows 2 graphs that chart the relationship between chronological and biological age, as measured by his methylation clock.
You can see that it spikes early in life, until about adolescence, then still quick until the early 20s, and then it flattens is follows chron. age very linearly.
I guess that at this point, you can make of it what you will. I think Horvath is honest is saying that we don’t quite know yet what conclusions to draw from all this. But very interesting correlations nevertheless.
Agreed. Thanks.
Hi Akshay
I was first introduced to IP 6 and the work of Dr. Shamsuddin through your very informative post regarding supplements which have been shown to prevent/treat cancer. You may recall that he has done extensive research showing that IP6 alters gene expression in cancer cells causing them to alter their differentiation from poorly differentiated to well differentiated cells which then behave in a normal manner.
So anyway, I have a pediatrician friend who was telling me a heartbreaking story of a 2 year old girl with an inoperable neuroblastoma wrapped around her spinal cord.
Recalling your post , I called Dr. Shamsuddin last week and told him about this case , and that I had read his book, and I can say that he was extremely helpful and concerned and offered to reach out to the family himself and said that he has indeed had success with this tumor type.
It may well be a stretch in this case, I really don’t know, but it’s nice to know that all of the work involved in putting together various posts and comments could result one day in saving the life of a child.
Certainly not a waste of time.
Paul you do some amazing stuff! What a nice gesture. I am so glad that Dr. Shamsuddin was so helpful and reached out to the family. I would love to hear about a happy ending to this story and if it so please try to get pictures of the child. I would like to treasure it. You are a kind and a good man Paul.
Thanks Akshay
But I’m just in a position to try some last ditch efforts, but this is all you and Shamsuddin. If nothing else it will give this family some much needed hope and I’m certain after speaking with him that he will be realistic regarding expectations when he talks to them.
Of course I agree with you Alan, but we have some excellent cancer centers here in Baltimore and they have already exhausted all of the standard treatments. Everyone understands that this is only an option of last resort.
A child with neuroblastona should be treated at Memorial Sloan Kettering not with food supplement. Just too crazy.
Hi Paul, what is the proposed mechanism of action of IP6?
Hi Mark
Shamsuddin wrote an entire book on IP 6 where he describes the mechanism in detail, but it’s been a year since I’ve read it and I wouldn’t want to do him an injustice by getting it wrong, so I’ll re-read it this week and let you know.
Paul Rivas,
On the other hand there are many other plants, besides ashwagandha (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4658772/), presenting in studies positive effects on the autonomic nervous system and/or in reduction in lactic acid production. Among these plants are quercetin, resveratrol, curcumin and crataegus oxyacantha (Hawthorn).
Thanks Carlos
I’ll check it out
Carlos
Can hawthorn be a digitalis substitute?
Paul, responding briefly to your question my feeling is that Hawthorn might offer some benefit for the prevention or in treatment of cancer. However, not to the point to guarantee that it has sufficient power to be a digitalis substitute.
As for eliminating sugar as a cancer treatment, I recently read that 2,4-Dinitrophenol (DNP) works against cancer using a similar mechanism. “In living cells, DNP acts as a proton ionophore, an agent that can shuttle protons (hydrogen cations) across biological membranes. It dissipates the proton gradient across mitochondria and chloroplast membranes, collapsing the proton motive force that the cell uses to produce most of its ATP chemical energy. Instead of producing ATP, the energy of the proton gradient is lost as heat.” The main side effect is body heat and sweating as well as lethargy from mitochondria not being able to produce ATP. Bodybuilders use DNP to burn fat off their bodies like a blowtorch.
Interesting. Are there any studies you can share Marcus?
Hi Aldebaran
The acarbose study is interesting. Do you have a reference for that one. When I started on rapamycin my blood sugar was at its usual 99. One month in I was down to 80. At 2 months it climbed to 100. At 10 weeks I really felt great on it and my sugar was up to 115 and it’s stayed there.I’m waiting on my insulin and IGF-1 levels, which are key in my mind. Also lost 15 lbs and exercise even more, yet my FBS stays at 115, it’s an interesting phenomenon.
Hi Paul,
Sorry I missed that post but I guess we have already discussed plenty about ACA. I guess that if your FBS raised but your insulin level dropped, it might not be that bad. Also, could the raise of FBS be a transient effect in relation to weight loss?
I think that I went from a certain degree of insulin sensitivity to some insulin resistance. It’s interesting. But I’m more concerned about insulin levels, IGF-1, and inflammation (crp), than I am about a rather arbitrary blood glucose. I mean according to the lab if my number is 99 then I’m ok, but if I’m 101 glucose then I should prepare my will.
I was taught long ago by a very wise and prominent mentor to treat the patient and not the lab.
I was working in a diabetic clinic and was very proud of myself for bringing down their blood glucose levels with insulin therapy but my mentor said to me, ” Good work Paul, , now you have a bunch of fat diabetics that you’ve created “.
Lesson learned
Interesting experience with diabetics! I have been told that the tolerance for mistakes is one of the strength of America. It encourages people to take risks. This is certainly true for high tech.
In the medical field we’re all making multiple mistakes every day. We just don’t know it yet
The great chess master Tartakover used to say that the winner is the one who make the last before one mistake.
Thanks for this. I believe I have throat cancer, so this may be my only post. I do think Melatonin can help with this and skin cancer though as it does seem to show some improvement in my symptoms.
Have fasted for three days and am not eating much now. Doing a hyperbaric session tomorrow.
Taking various other B Vitamins, but don’t think they are having much effect.
Am seeing a Doctor within the next two weeks, but am not expecting a hopeful prognosis at all.
Or maybe ahem, it was just reflux. As someone said at work – if you hear hooves behind you think horses. Not Zebras*
* unless you are running on the serengeti