
Cell biologists are within striking distance of “partial reprogramming”. Already, technology has arrived to turn an old cell into a young cell in a Petri dish, and researchers (Turn.bio) are looking intensely for ways to safely rejuvenate cells within a living body. Is this the breakthrough that we in the human rejuvenation movement have been waiting for, or is it a sideshow?
Partial Reprogramming
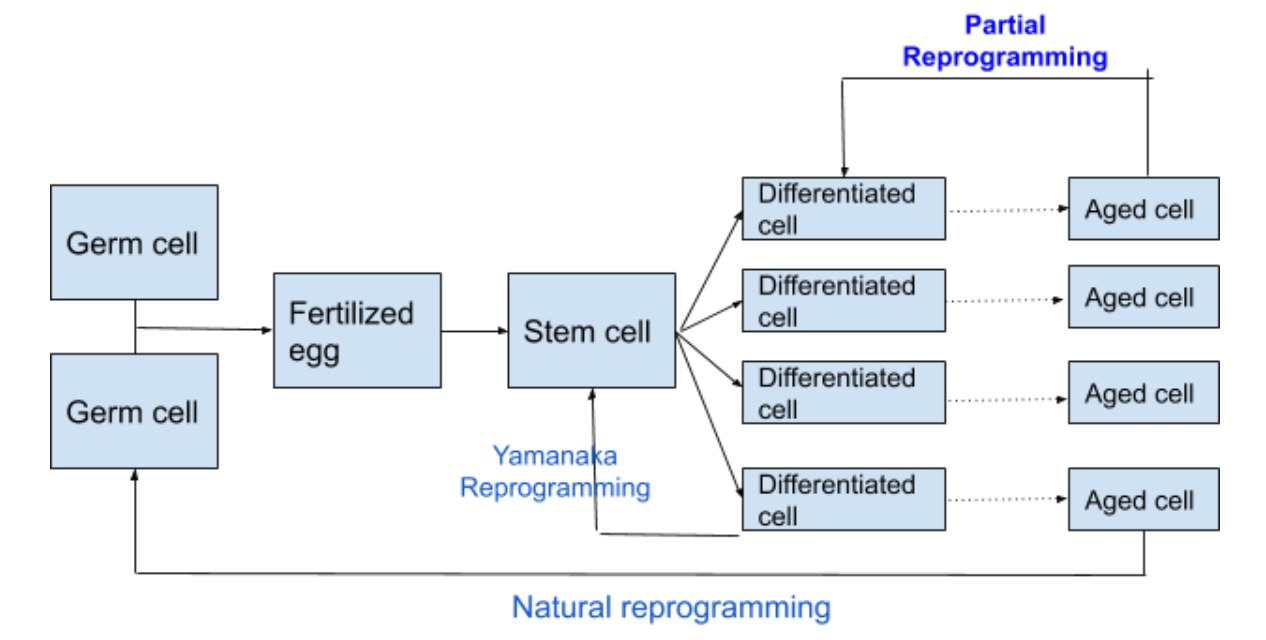
In nature, aging is part of a one-way street. A germ cell becomes a stem cell becomes a differentiated cell, and then the differentiated cell grows old.
 In the course of nature, cells change their epigenetic state from left to right. Nature must have a mechanism for resetting the cellular aging clock, going all the way back to the left. If this didn’t exist, then all cells would be on a one-way path to extinction. At some point in the life cycle, nature needs to take a mature cell and turn it into a germ cell (sperm or egg). But, in the process, epigenetic programming is wiped clean. Two things happen simultaneously: memory of the cell’s functional differentiation is lost, so it becomes again a pluripotent stem cell; and the age of the cell is reset to zero.
In the course of nature, cells change their epigenetic state from left to right. Nature must have a mechanism for resetting the cellular aging clock, going all the way back to the left. If this didn’t exist, then all cells would be on a one-way path to extinction. At some point in the life cycle, nature needs to take a mature cell and turn it into a germ cell (sperm or egg). But, in the process, epigenetic programming is wiped clean. Two things happen simultaneously: memory of the cell’s functional differentiation is lost, so it becomes again a pluripotent stem cell; and the age of the cell is reset to zero.
It never happens in nature that the cell’s epigenetic age is reset to zero, without also erasing the cell’s functional identity. Nature has no need for this process. But for cellular rejuvenation, this is what we would like to be able to do. If all the cells in your bones became young again, you might lose the calcification and brittleness of old bones and regain the springy resilience of a 10-year-old. But if all the cells in your bones became stem cells, your bones would lose their structural integrity and your body would collapse like a mass of jelly.
In theory, we might learn enough about hundreds of epigenetic changes that take place with age, and use CRISPR or analogous process to reset each one of them individually. This would be cellular rejuvenation “by hand”. If we are really, really lucky, then this Herculean biochemical task might be avoided by some accidental pathway by which the cell resets these hundreds of epigenetic markers on command. But we have no reason to expect that a mechanism exists to do this, because in the normal course of a life cycle, nature has no need for it.
Thirteen years ago, Yamanaka [2006] found that differentiated cells (specifically skin cells) could be induced to revert to stem cells by exposing them to just 4 proteins, which have come to be known by their initials as OSKM, the Yamanaka Factors. This was akin to what nature does, resetting the cellular age and erasing the cell’s function. Then, three years ago, a study from Juan Carlos Belmonte at the Salk Institute gave us hope that de-aging a cell might be possible without loss of its identity. They used the same OSKM, but exposed the cells for just a few days, then turned off the exposure. They reported that the cells were made younger without erasing their function. Mice with the rejuvenated cells lived longer. This was a proof of principle, but there were big caveats. First, they worked with progeria mice, genetically programmed to age unnaturally fast. Second, the mice were genetically prepared with OSKM grafted into their DNA, and pre-coded with a chemical switch so that OSKM could be turned on and off at will by injecting the mice with doxycycline. For mice that are not genetically modified before birth (or for normal people), delivery of OSKM to individual cells and timing that delivery poses a substantial challenge.
Then, in a preprint posted to BioRxiv just this spring, Vittorio Sebastiano and his Stanford group took another step forward. They added two more ingredients to the Yamanaka recipe (OSKMLN) and succeeded in rejuvenating human fibroblasts in cell culture, as reported by the methylation age of the cells. This experiment had neither of the two limitations of the Belmonte group, and it was human cells rather than mouse — three steps forward. But it was done in vitro only — one big step backward.
Turn.bio is a biotech startup that is seeking to develop and capitalize on the technology. Steve Hill of the Life Extension Advocacy Foundation (LEAF) interviewed Sebastiano about his discovery and the path forward. Hill provides more background in this article. Over at FightAging!, Reason reviewed the subject.
Is epigenetic reprogramming a driver of aging, or a response to cellular damage?
Hill asked Sebastiano this question, and he hedged in his response:
My personal opinion is that I can’t really decide whether the epigenetic changes are the cause or the consequence. I cannot decide what theory is right in the sense that some people suggest it’s a developmental program of aging and some people say it’s a consequence of damage accumulating. What I really care about, at the end of the day, is that, regardless, epigenetic changes explain aging. The epigenetic changes are what, at the nuclear level, triggers this dysfunctionality of the cell.
— Vittorio Sebastiano
The logic in this answer is incoherent. I suspect that Sebastiano is not confused, but he knows what he has to say to keep his funding flowing, and to keep from being distracted by philosophical arguments. There is a prejudice in the field that he has chosen to skirt, rather than confront it head-on. Look at his last sentence, “The epigenetic changes are what, at the nuclear level, triggers this dysfunctionality of the cell.” He recognizes that altering the epigenetic program is going to make the cell younger, but he avoids saying that the body has arranged the epigenetics to make the cell older.
Aging as an epigenetic program
The core truth here is that alteration of gene expression is the way the body functions. Gene expression is different from cell to cell, from tissue to tissue. The way the body changes its strategies from minute to minute and also from decade to decade–also gene expression. Epigenetics = gene expression is the heart of the way the body’s metabolism and the core of the developmental program by which we grow arms and legs and bones and muscles. It is also the core of the aging program, but you can run afoul of funders, decision-makers, journal editors and other gatekeepers if you say so. Better not to say so.
We know the cells of nearly every tissue are epigenetically reprogrammed as we get older. Is the purpose of this reprogramming to resist the damage, which is the primary cause of aging? (standard theory) Or are the epigenetic changes implemented as a self-destructive program for the express purpose of weakening and then killing the body? (programmed aging theory, to which I subscribe)
This is no abstract question for theorists–it has fundamental implications for practical anti-aging research. If the epigenetic changes are there to resist aging as best the body knows how, then we shouldn’t be tampering with them. But if the epigenetic changes exist only to create damage and stymie the cell’s repair mechanisms, then restoring the epigenetic program of the cell to a younger state looks like a promising anti-aging strategy.
Reason on Cancer
The response at FightAging! to Sebastiano’s experiments with cellular rejuvenation starts with a presumption that this kind of intervention must raise the risk of cancer. Where does this presumption come from? His thinking is based on general principles of evolutionary theory. Theory says that the body is trying to live as long as possible, and if the body has made the decision to permit cells to senesce, it must be from a self-interested calculation that it is better to allow certain but slow death in the guise of cellular senescence than it is to risk the possibility of near-term death from cancer.
I believe the evolutionary theory is wrong, and if so, there is no a priori reason to think that cellular rejuvenation will increase cancer risk. In fact, we might hope that cancer risk decreases, as the body’s immune system is restored to a younger state and systemic inflammation is quelled. (Of course, we will still want to experiment with animals and then humans to assure ourselves that the treatment does not increase cancer risk.)
I have staked my professional career on the theory that aging is programmed self-destruction, that the body is not trying to live as long as possible, but rather is aiming for a predictable lifespan, and if we thwart that program, we won’t have hell to pay.*
Clear logic of programmed aging
Aging is an epigenetic program, honed by natural selection for the sake of the community over the individual. The one-line proof is that genes regulating aging have been preserved in the genome since we were descended from single-celled ancestors 1 billion years ago. A longer version is in this blog five years ago, and the 300-page version is in my book.
Once you accept that aging is programmed, it follows that aging must be coordinated system-wide. We can look for one or several clock mechanisms, and for signals that transmit the age-state of the body through (almost certainly) the blood plasma. The quickest path to rejuvenation technology is not “repair of damage” — a daunting challenge of bioengineering — but only a modification of the signaling environment, or, perhaps, direct manipulation of the body’s aging clocks.
Cellular Rejuvenation: The Path Ahead
When the treatment matures, what will be our strategy for the body as a whole? Is there a central clock (perhaps in the hypothalamus, a neuroendocrine region of the brain) where the treatment must be targeted, after which the rejuvenation signal will be transmitted to the body without further intervention? Or would we have to reprogram every cell in the body?
What about inflammation? Presumably, systemic inflammation is controlled by signal molecules that will revert to youthful levels after reprogramming.
What about arterial plaques? Will they be cleared up by a rejuvenated metabolism? Same question for beta amyloid in the brain?
What about oxidative damage? Would the body know how to pick up the ball that it dropped when we were much younger? What about cross-linking? Accumulation of lipfuscin?
At times like these, I’m shaken awake to realize how little I really know about the aging metabolism, and the signal transduction that drives it.
————-
Perspective
For me, this is a case where the technology has gotten ahead of the science.
The big picture is that from the 1950s, evolutionary biologists have handed the medical researcher a mistaken framework. Medical researchers have done their best to ignore the theory and forge ahead with a practical program that addresses the changes that are observed to take place with aging. This agnosticism is a lot better than sticking dogmatically to a flawed theory.
But we could do so much better — we will do so much better — when we embrace the correct theory. A clear theoretical framework will be extremely helpful in guiding lab experiments toward the most important questions.
Here’s what I mean, specifically: Evolutionary theory offers the clear message: the body cannot have organized programs of self-destruction. This implies that aging is a disorganized process. It must be damage. It must be random and it must be local. It makes sense to learn about the cellular biology of aging, and develop ways to heal the aging cell. Aging will be remediated from the bottom up.
But the theory is wrong. In fact, aging is coordinated systemically. It is a top-down process, directed by signal molecule in the blood. The most efficient way to remediate aging is to study the signaling mechanism, to understand it well enough that we can alter the signaling environment, telling the body that it is young. We don’t have to repair damage in every damn cell in the body. All we have to do is to re-adjust the levels of hormones and transcription factors that circulate in the blood to youthful levels.
Once we think this way, it is obvious where the focus of our research ought to be.
- We need to understand how the system is coordinated. It is not yet known whether the clock that controls aging is in a specific location, probably the hypothalamus deep in the center of the brain, or whether the clock operates as a consensus among many distributed sites (e.g., telomere lengths and methylation states in many tissues). In this latter picture, the transcription factors that circulate in the blood and dictate epigenetic state are generated throughout the body, contributed by every cell in every tissue.
- Even more important, we need to catalog the thousands of signal molecules in the blood, proportions of which change with age. It is likely that some of these are more important than others, and if these few are reset to youthful proportions, the rest will follow. How many? Is there a manageable list of signal molecules that can be re-balanced in the bloodstream, and it will reprogram all the rest? Or must we manipulate hundreds of separate hormone levels in order to turn back the aging clock? The answer is yet unknown. A related question: How long must the blood levels of these compounds be artificially maintained before the body is reprogrammed to a youthful state, and the intervention is no longer necessary? We might imagine people lined up for a once-every-decade trip to the rejuvenation clinic with an IV drip for two days. But if the treatment has to be sustained for months at a time, it will be prohibitively expensive, uncomfortable, and disruptive.
Here’s an example that comes from this kind of thinking — an experiment we might start with: Take a sample of blood plasma from an artery going into the brain of a young mouse (or human), and catalog the proteins and RNAs. Do the same with the blood plasma emerging from the brain. “Subtract” the two profiles with a computer comparison to see which elements are changed. Any significant differences might tentatively be imputed to the hypothetical hypothalamic clock. Repeat the two measurements and the differencing with an old mouse. The difference of the differences is a good first guess as to what molecules in the blood control aging.
Back to Cellular Rejuvenation and Partial Reprogramming
Cellular rejuvenation may turn out to be a crucial technology for us to master, or it may be something we don’t have to understand in detail, because the body does this by itself once we rebalance the signal molecules in the blood. Or — a third possibility — it may be that cellular rejuvenation in the hypothalamus is sufficient to reset the body’s global aging clock. We could be addressing these questions experimentally.
Discover more from Josh Mitteldorf
Subscribe to get the latest posts sent to your email.
A few questions:
1.) Would reverting a cell’s state back to an earlier state erase any epigenetic changes that have accumulated over the cells life?
2.) Is this something that could potentially be DIY? I fear that if we leave this in the hands of the FDA we are looking at 10 to 20 years for access, by which time some of us reading this blog will be quite dead.
pyridoxine and l carnosine interfere with glycation,a worse problem than oxidation,slowing the aging process whereas panax ginseng increases hair growth and telomeres as does tocotrienols as well as myriad other benefits as well as heart and stroke prevention.Tocotrienols and pomegranate remove arterial plaque and soften endothelial cells.High absorbable vitamin b,especially b-6,b-12 and folate maintain telomeres.Quercetin and bromelain remove senescent calls allowing for healthy cells to predominate.These are all highly anti-cancer while only improving healthy cells age slowly.
Lobsters are one of the few species that are immortal and become stronger and more fertile with age.They can live more than 100 years.How?Their food chain consists of other animals,crab,mussel,starfish and others whose food chain is all dependent on algae.Algae grows for many years in the sea with only the sun to cause damage.It develops an immune shield of red colored astaxanthin which has been found to be the most powerful antioxident discovered.From salmon to crab to krill to pink flamingos, astaxanthin is in each creature.The largest animal on earth,the blue whale,eats mostly krill.The lobster has ever increasing telomeres throughout its life.Astaxanthin has shown recently to prevent multiple sclerosis and reverse it.No doctor will prescribe it as they are ignorant and arrogant only peddling synthetic FDA poison and feeling proud to do their job.
Wonderful article –
Dear Josh,
Just wanted to point out that the coordinated, system-wide aging process might not only be controlled by signals in the blood, but also by the vegetative nervous system. See
PMIDs 29932493, and 30303708, or this article on ScienceDaily: https://www.sciencedaily.com/releases/2018/10/181017141033.htm
Electric communication through the body is a large subject, almost completely ignored. It may turn out to be a sleeper!
Dear Josh, since you have mentioned to the lipofuscin, when your occupations allow, you could give us your expert opinion on DMA or dimethylglyoxal-apigenin: Apigenin and its methylglyoxal-adduct inhibit advanced glycation end products-induced oxidative stress and inflammation in endothelial cells,
here it is in full doc https://www.hindawi.com/journals/omcl/2019/9026456/
Best regards! A.
Josh,
I think you are right that cellular reprogramming is the holy grail of anti-aging in that it would repair all aging damage regardless of cause. In worse case scenario, where aging is due to genomic DNA damage, cellular reprogramming combined with the stem-cell targeting gene therapy developed at Harvard recently would effectively solve the problem.
Aubrey de Grey, who is at the competitor to Turn.bio, AgeX (both companies are pursuing cellular reprogramming and are, thus, competitors) has become more optimistic on near-term (less than 20 years) breakthroughs in aging. Some of his optimism is based on the fact that lots of money is now flowing into the field. However, I also believe some of his optimism is based on the recent cellular reprogramming work as well.
The key point to keep in mind about biology in general is its inherent dynamism and regenerative nature. This is why the “civil engineering” paradigm of medicine and of aging in particular is so wrong.
…This is why the “civil engineering” paradigm of medicine and of aging in particular is so wrong….
Yes! The automobile garage, “damage repair” paradigm is so very wrong…
‘Unreasonable’, one might even say….
I agree. I think this stems from the misleading metaphor of seeing the body as a machine. Although cells group together to form tissue types and organs, at its core, an animal is a community of clonal cells selflessly cooperating for the survival of the germ line, a subset of that community of ‘sisters’. This is almost a form of group selection if you think about it.
Each *is* is a machine. Capable of self-repair, replication no less, and often of autonomously seeking nutrients. We don’t quite have any machine that does that, but I think the metaphor would be closer than that for the whole body.
Although the Interplay between those billions of microscopic machines that make *us* is key, the question as to ageing lies in determining which is the root cause: the mechanisms inside the cells, or the interplay (extracellular communication) between the cells. Both are important, to be sure, but I would say internal cell mechanisms are upstream.
In regards to the central question of if epigenetic changes are a cause of or a reaction to aging. My interpretation of parabiosis experiment results and observations of young blood transfusions would seem to strongly support the theory that epigenetic changes are causal and the signaling is at least partially resident in plasma. But perhaps those observations do not as strongly support that conclusion as I would like to think. What else could be a reasonable alternative to explain the observations from parabiosis and young blood transfusions?
I wonder whether the signal molecules in the blood driving aging is due to excess protein in the diet.
Ron Mignery, a PhD that works in the Boston area, has a free online book called the Protein Cycling Diet that outlines his thesis. I don’t know if he still is, but he had been consuming a very low protein diet (based on percentage of calories from protein).
https://proteincyclingdiet.wordpress.com/article/protein-cycling-diet-2s3nmvrwklbxs-1/
http://proteincycling.blogspot.com/
And Ron Rosedale touches on the adverse effect to mTOR from too much protein in the diet. So if I had to guess, I would say that excess protein in the diet promotes the type of molecule signalling that drives the aging process.
On these global clock signals that coordinate the aging program – it would seem that if you had a set of mutations that took out all the signals you might have an organism that is genetically immortal. Since we don’t have known examples of people that don’t age, if you could estimate the mutation rate per signal you might be able to bound the number of different signal paths to produce no non aging examples in the observation time.
Of course there would be a lot of hand waving involved, but it might give you a reasonable range of paths that you might expect.
Also, given the expectation of multiple independent global clock signals, I wonder if the examples of very long lived individuals might be shown to have some of these paths disabled and they only get aged out by those that remain.
The fact that there are multiple signal paths would seem to infer that evolution really don’t want any immortal organisms and therefore has built in sufficient redundancy to insure that never occurs.
That seems like a darn good idea to me Michael. Of course, I am a layman.
I love your work on this blog very much.
In fact, aging is coordinated systemically. It is a top-down process, directed by signal molecule in the blood. The most efficient way to remediate aging is to study the signaling mechanism, to understand it well enough that we can alter the signaling environment, telling the body that it is young. We don’t have to repair damage in every damn cell in the body. All we have to do is to re-adjust the levels of hormones and transcription factors that circulate in the blood to youthful levels.
surely, this is of direct relevance:
https://joshmitteldorf.scienceblog.com/2019/02/05/rumors-of-age-reversal-the-plasma-fraction-cure/
Transgenerational epigenetic inheritance studies demonstrate that epigenetic programming is NOT wiped clean in germ cells. Understanding these mechanisms may help discover similar mechanisms in somatic cells.
Dr. Michael Skinner’s lab at Washington State University is a leader, with studies like “Transgenerational sperm DNA methylation epimutation developmental origins following ancestral vinclozolin exposure”
https://www.tandfonline.com/doi/pdf/10.1080/15592294.2019.1614417?needAccess=true
The studies main hypotheses were:
“Following fertilization, the hypothesis is that the transgenerational epimutations modify early embryonic transcriptomes and epigenomes to re-establish the cascade for the next generation.
As the individual develops, all somatic cells have altered epigenomes and transcriptomes to promote disease susceptibility later in life.“
Are there any known mechanisms to erase epigenetic programming?
Hi Josh
David Sinclair has a book coming out September 10, “Lifespan, the Revolutionary Science of Why We Age and Why We Don’t Have To.” I read a preprint and taped a
long interview with him on July 10th. I have injected some of the footage into my new film, “To Age or Not To Age – Transforming the Human Condition” – In short,
David has enunciated and underlying theory for aging, something that accounts for all the hallmarks of aging. It’s an informational theory of aging, “aging is simply the loss of information” – in a series of experiments that have lasted 10 years, they have restored sight in a mouse who they made blind by selectively using 3 of the Yamanaka factors.
He compares epigenetic aging to accumulated scratches on a CD, cells essentially lose their identity across the board. and it’s caused by the survival circuit. He was able to speed up aging in a mouse by creating random DNA breaks, creating more noise more scratches. And he was able to get rid of scratches.
Another way of looking at it might be that somatic cells have no way of remembering exactly what their epigenetic state should be – somatic cells are just progenitor cells whose consistent signalling eventually made the cells lose their previous plasticity; likewise progenitor cells lost the epeigenetic plasticity of stem cells due to the repetitious tasks they were assigned. With the right signalling it’s not surprising you can revert somatic cells to progenitors (ROCK inhibition) and even to pluripotent stem cells (OSKM).
Robert, that doesn’t seem to explain the cause of the scratches, which I think is what Josh is getting at with his theory of programmed aging through group selection effects. Does Sinclair’s book go into this?
yes David does. “as we age, threats to survival such as broken DNA, activate the survival circuit and rejigger the epigenome in small ways…over time cells progressively lose their original identity, eventually transforming into zombielike senescent cells in old tissues.” Personally, whether we try to torture the word program is not the issue. The key is that the cell retain the information of how to be young. According too Sinclair, that’s why cloning produces a young animal even though the animal is derived from an old animal.
but that’s just restating the idea that scratches accumulate. Why do they accumulate under Sinclair’s theory? Just wear and tear basically?
I think what he means is that DNA has to be unrolled and demethylated for DNA repair (which is highly efficient), but after repair the remethylation is not as efficient, leading to errors over time; the more DNA repair required, the faster the accumulation of remethylation errors. It is like I said before, somatic cells get gradually further from their progenitors via this gradual drift. There is also probably intentional methylation changes as the cell adapts to other forms of aging changes, for example loss of proliferative ability due to telomere attrition in tissues.
DNA methylation can be lost by oxidative stress, and can be acuired by errors of the methyltransferase complexes. Also hisetine depletion might play a role. When the DNA is synthetized a different type of histone subunit is used than when the histone is replaced.
Although turn.bio’s work is interesting, I think better approaches will be found. The whole idea of partial reprogramming seems attractive for in Vivo, but they are mainly focussed on in vitro work, and it now appears induced pluripotent stem cells added by IV are in fact very safe anyway and likely highly efficacious. So this may be he way to go.
It seems you also believe this Josh, as you start the blog of talking about cellular reprogramming, but end up advocating a more systemic approach.
I think you may well be right that a group selection for aging may occur. Regarding the risk of cancer with regeneration, most regenerative species are highly resistant to cancer. So it my not be the trade-off we’ve always been told it was. So long as the karyotype can be protected, cancer can be prevented.
Excellent summary. I’m pretty convinced by your arguments for programmed aging but look forward to more evidence along the lines of what you sketch here. Any papers you can suggest on the role of the hypothalamus in programmed aging?
Here’s a 2018 review “Role of hypothalamus in aging and its underlying cellular mechanisms”
https://www.sciencedirect.com/science/article/pii/S0047637418300502
The hypothalamus is a brain structure that lacks feedback mechanisms for several of its activities. This structure develops shortly after conception and has an active prenatal role. The hypothalamus plays its part in getting us developed and ready to reproduce, with certain feedback loops being evolutionarily unnecessary.
The hypothalamus perfectly illustrates the Blagosklonny point of “When these programs are completed, they are not switched off.” Hypothalamic activity not winding down when its developmental role is over shouldn’t be interpreted to construe a role that has some other meaning or purpose.
Also, Whittemore and Blasco’s recent paper showing a tight correlation between telomere attrition rates and average species longevity suggests to me that this may well be, as Fossel has suggested for years now, the central command post for programmed aging. Thoughts on this paper?
Great paper. It’s remarkable how good Michael Fossel’s predictions have been. For example he’s said for many years that mice age the same as humans, and it’s the rate of shortening of their telomeres that matters, not absolute length.
Josh: not sure why the aging “establishment” is so adamant that aging can’t possibly be programmed given that there are clear examples to the contrary. For example, fish physiologists have long known that aging is programmed in Pacific salmon. Adult salmon age rapidly after entering fresh water to breed and die within a few weeks of spawning. The dramatic physiological changes that take place have been shown to be hormonal driven and can be reversed, at least initially, by returning the fish to sea water. I don’t think anyone has looked at what is happening at the epigenetic level. There’s a large literature on the ecological and evolutionary significance of this life strategy. Paraphrased, it is that the parents are making the ultimate sacrifice for their offspring by adding essential nutrients in the form of their decomposed bodies to the nutrient poor freshwater ecosystems where the young salmon spend their first few years. Pacific salmon seem like an interesting model to test aging theories and I’m surprised that no one has done much work in this area, at least recently. Most of the physiological literature dates from the 1950s to 1970s.
https://www.ncbi.nlm.nih.gov/pubmed/16212264
@Ian: and since this is biology, here’s a paper that claims that the paper you cited is wrong:
https://www.ncbi.nlm.nih.gov/pubmed/21137209
i’ve seen mentions that activating telomerase gene (demethylating it) also is resetting methylation of the whole dna, as the site controlling methylation is located next to it, but I don’t remember the source. Using scuttelaria baicalensis (which, after TAM818 and TA65, has the strongest telomerase activation ) ungreys some gray hair, anyway (underlying mechanism not known so far).
Can you give more details on ungraying? I’ve been trying to find good info on this for a while but all I’ve found so far is interesting repigmentation effects from some immunotherapy drugs.
Where do you find that Chinese Skullcap (scutellaria baicalensis) promotes telomerase? That is completely opposite of what I find!
Baicalin, apart from being water solluble and activating p53, is a twin to silibinin (main constituent of sylimarin) which is fat solluble. Sylibinin was long believed to inhibit telomerase (after studies in many cancer cell lines) and was placed in every table as ‘telomerase inhibitor’. Until the day the assay was made on healthy liver cells, when it was discovered that “Silymarin increased telomerase activity 3-fold”. Baicalin cures liver fibrosis as effectively as sylimarin. Baicalin also has a strong anti-photoaging properties (including protecting telomeres), but is reported to not cause telomerase in epidermis cells (but the study did not report how many skin cells divided in the sample, and as photoaging is not activating cells divisions and telomerase is produced only on division, so this particular study is still inconclusive on telomerase expression in this kind of cell. I also have seen a few studies of baicalin on different cancer lines but I failed to find so far a study on telomerase expression in healthy liver cell lines…
I would say that if a substance is identitical in most of it’s properties to a strong telomerase activator, it would be likely that it works the same way with it too. The subject is a very interesting matter to further study to clarify how much baicalin properpties are twin to sylibin in this respect and how much are based on its’ p53 properties (as a senolytic and anticancer path). Anyway, the case of sylibinin says that tables of “telomerase inhibitors” missing the exact cell line where this inhibition takes place are pretty useless :< cause string 'inhibitor' can be at the same time strong activator.
[*links omitted since they block message form appearing on site*]
I remember in 2015, the news that Japanese scientists, by the addition of glycine for 10 days to an elderly Human cell line, restored damage of aging to the cells. It made news with newspapers speculating that the elderly could get a new lease of life by supplementing with glycine. No information was given as to what amount of glycine would replicate the research. I have since then taken daily supplementation of 1000mg, so although what an optium amount might be is unknown, I take the longer term view that one glycine tab a day is 364 a year, so 364 on a yearly basis must be having some effect.
Although in-vitro, there seems to be some concern regarding glycine supplementation in regards to prostate cancer.
https://www.ncbi.nlm.nih.gov/pubmed/26847870
Sarcosine-related amino acids (glycine, dimethylglycine, and sarcosine) can exceptionally affect the behavior of benign and malignant prostate cells.
Does anyone know what drug New Scientist is talking about?
https://www.newscientist.com/article/2211927-exclusive-can-a-supplement-slow-the-natural-processes-of-ageing/
So I got my hands on the article. It seems interesting.
Florida based company called Ponce De Leon Health (PDLH) has released a supplement which they say have shown promising results on mice. It’s called Rejuvant and contains alphaketoglutarate (AKG) in combination with vitamin A for men and D3 for women. They say there are evidence AKG targets senescent cells.
Quite expensive stuff, and I don’t know anything about this company. The homepage does seem sensationalist. Do you guys know something?
AKG is not expensive, and both vit A and D are very cheap.
Places to buy
Alright, so the drug is not that interesting ?!
Is there any science that links alphaketoglutarate with increased lifespan aside from the study on nematodes a few years ago?
Unless it shows effectiveness in higher mammals (at least mice) I don’t get very excited.
Michael, in the article it says increased lifespan have been observed in mice, and also healthier life. I’m just quoting the article, I have no knowledge of this myself. The improvements, in theory, for humans, is 5 more healthy years. The experiments on mice is under review for publication in a journal.
The company is preparing a 80-person trial to test for safety. and also look for epigenetic changes.
Agreed. Fossel doesn’t seem to get the attention he deserves. Or the funding.
after we are born , the body grows in a coordinated fashion . the signalling for the growth must be the factors in the plasma i.e certain factors must be signalling to the various organ systems , therefore certain factors are common to growth taking place. this growth has to stop somewhere, it cannot go on indefinitely. I think a final morphological feature introduces growth inhibiting factors to counter growth inducing factors. these growth inhibiting factors are what i feel the cause of aging. aging could be the aftereffect of growth tapering off and stopping
It’s interesting that the species that don’t seem to age, don’t seem to stop growing. And indeed we know from bacterial ‘aging’ that cells that divide can divide any accumulated ‘damage’, asymmetrically if need be, such that in stressful circumstances atleast one daughter cell line will survive. But also the species that continue to grow have the proliferative ability via telomere maintenance for their cells to continue to divide. Humans do not have this ability, but have a long life purely through well controlled metabolism. If we wish to live longer greater cell cleaning, i.e. autophagy, lower oxidative stress, etc., can get us only so far – what we really need is improved cellular proliferative capability. The fact that ‘ageless’ animals have this ability suggests this is a viable path to longer life for humans (who have very short telomeres, albeit with a slow rate of loss).
young plasma can be a viable growth inducing mechanism, and repeated cycles of young plasma factors may be the holy grail but the biggest problem is there is no big money in young plasma. Aging as opposed to cancer is not a disease with myriad variants, it most likely has a similar cause and a singular path to solving it. If young plasma is the cure, the question is can so many companies profit from it
Maybe young plasma, with all the right signalling molecules could reset the age of cells around the body. Assuming this can be done, or the most important factors isolated, concentrated and applied instead (I personally think unaltered plasma will be too weak), the next question, as Josh asks, how long would you have to apply such treatment? My guess is the time period would be substantial, and the older you are the longer the treatment would take. Reaching areas of the body with little blood supply could also be problematic, depending on the molecular size of the relevant signalling molecules. Simple factors in the blood are certainly a holy grail of anti aging interventions, not least because they could potentially shortcut hugely expensive alternatives like gene therapy. The problem you accurately state is that it might not be half so profitable for its developers. After all, once the important factors are known, which presumably are ‘natural’, what is there to patent?
I would assume that the specific combination of the natural factors needed to achieve the desired outcome would be patentable. And since you’re almost certainly going to have to administer this IV, there’s not much opportunity of someone making some sort of supplement to circumvent you. I think since you’d require an FDA drug approval and you’d have a patent on the combination you’d have a route to make a substantial ROI.
Of course, we used to have universities doing this sort of basic research with the idea of serving humanity rather than having an eye on making enormous profits, but that seems to have gone away as they’ve all gotten focused on drug development in the last 30 years or so.
Maybe, but presumably the ‘combination’ of factors is present already in young people. Plus once those factors are known and manufactureable, i.e. not requiring blood, it wouldn’t be overly difficult to set up an IV clinic. Bottom line, solving aging this way might not make the expected billions for its pioneers. I hope they can live with that.
I picture the potential for more of a dialysis machine than an IV drip. My understanding from current young blood transfusion observations is that removal of the old is as important as introduction of the young. Therefore, if it is possible, there might be more of a need to “scrub” the plasma instead of just adding new signaling molecules. My guess would be that there would be plenty of money making opportunity in such a tech heavy setup that entrepreneurial motivation is likely not an issue.
Love your blog Josh. Isn’t Dr Sinclair’s latest study exactly what you are talking about?
Abstract
Ageing is a degenerative process leading to tissue dysfunction and death. A proposed cause of ageing is the accumulation of epigenetic noise, which disrupts youthful gene expression patterns that are required for cells to function optimally and recover from damage1–3. Changes to DNA methylation patterns over time form the basis of an ‘ageing clock’4, 5, but whether old individuals retain information to reset the clock and, if so, whether this would improve tissue function is not known. Of all the tissues in the body, the central nervous system (CNS) is one of the first to lose regenerative capacity6, 7. Using the eye as a model tissue, we show that expression of Oct4, Sox2, and Klf4 genes (OSK) in mice resets youthful gene expression patterns and the DNA methylation age of retinal ganglion cells, promotes axon regeneration after optic nerve crush injury, and restores vision in a mouse model of glaucoma and in normal old mice. This process, which we call recovery of information via epigenetic reprogramming or REVIVER, requires the DNA demethylases Tet1 and Tet2, indicating that DNA methylation patterns don’t just indicate age, they participate in ageing. Thus, old tissues retain a faithful record of youthful epigenetic information that can be accessed for functional age reversal.
https://www.biorxiv.org/content/10.1101/710210v1.full
Only half is “epigenetic noise”. The other half is an epigenetic program. If it weren’t for directed, non-random epigenetic change, the methylation clocks would not be possible. In fact, methylation clocks are the most accurate age measures we know. This alone is proof that aging is–at least in part–an epigenetic program.
I’m pretty sure a clock is possible with random noise, just as an atomic clock is super accurate despite the random timing of an individual nucleus’ decay. This would be analogous to the fact that the methylation clock is effectively looking at an average of the methylation state across the genome, compared to a reference at different ages.
As for the programmed element of epigenetic changes, we don’t yet know if this is causal – it could be a reaction of the nuclear program to other problems.
I agree the program might be simply a gradual abandonment of the repair program driven by the accumulation of damage.
I would speculate that the program might be a gradual abandonment of the growth program, which is gradually inhibited to stop growth.
Mark – having interviewed Sinlcair for 90 minutes 3 weeks ago, you are very close to his thinking; he
accelerated aging in mice by creating random DNA
breaks at a 50% higher incidence, they were not
done in any particular area. Those mice grew old predictably faster, across the board, not b because this or that organ system was tampered with.
Robert, when and where is your interview published?
You could build a clock on nothing but a purely random process like radioactive decay. It would have poor short term accuracy (seconds and less) but good long term accuracy (minutes, hours, days, and longer).
Certainly whatever clock is running the aging process doesn’t care about time frames less than something on the order of a year I’d bet.
There are 25,000 CpG islands on the human genome, yet only a few hundred, depending on the ‘clock’, appear to track age. For this process to be random the affected islands should happen to be more ‘prone’ to lose or gain methylation at a factor of 50 or 100 compared to the ones that don’t. And not just one or the other for each case, but a particular state for each case and age.
Cells also seem also capable of selectively back tracking this process when exposed to OSKM factors.
This really looks like a highly controlled process. And it is no surprise if we think of how important gene expression is to development.
Ultimately it could be a random process, the real question is, is it causal? Really, we could also say evolution is largely random, expect perhaps in sexual selection. There is no one in charge, yet it behaves as if in an apparent directed manner.
You make a good point Adrian. Though you can clearly build a clock from random changes, these changes don’t look random.
But I wonder. You might be able to build alternative clocks using other CPG islands with no overlap with current clocks. And there is no obvious connection to gene expression using current clocks. And yet we know the OSKM process extends telomeres (HTERT gene) and restores mitochondrial function (GCAT and SHMT2 genes), allowing cells to start over. So it is a conundrum.
Is this methylation a universal metabolic process that is unconnected to the functional improvements caused by OSKM proteins or to the degradation of aging? It’s seems a stretch to say so. But there must be some common pathway to the alternative clocks to be casual to aging – or maybe they are only correlated to aging, caused by the same process that drives aging, but none are actually upstream.
Evolution only looks directed in hindsight. You forget all the species that weren’t viable.
I’ve looked directly at methylation data vs age. There are many sites that appear to be losing methylation focus, with random changes.
But the sites that work best for constructing a clock are changing steadily with age. There are many thousands such, and I was looking at less than 1% of the whole genome. For some methylation increases linearly with age (after maturity) and for others methylation decreases linearly with age (after puberty). These sites contribute to the best clocks.
Hi Josh.
So certain sites used in the best aging clocks tend to gain methylation, and others tend to de-methylation?
This sounds like a well ordered process.
Maybe it is related to the process of re establishing epigenetic control occurring after DNA repair. This would explain its seeming link to metabolism, which is probably the main cause of DNA damage in cells.
Adrian, I am not sure about the number of CPG islands, but Illumina 450k chips check for methylation changes across 450 000 CpG sites. And yet Illumina 450k covers only a fraction of potential methylation sites around 30 million CpG dinucleotides in the human genome. The IIllumina sites were selected as known/putative promoters, enhancers, gene bodies etc.
Please note that Horvaths work is based on the more restrictive Illumina 27k chip, because there had been more datasets with that older version of the chip. Even when they have measurement data with Illumina 450k they convert it back to Illumina 27k.
Moreover Horvaths clock and other epigenetic clocks are just subsets of an infinite number of possible clocks. It really depends on your train and test set which sites are eventually chosen as a basis of your clock model.
I think no one really looked into the whole methylome inclusive of noncoding regions, telomeres, centromeres, etc. It may happen that epigenetic changes with aging have nothing to do with gene expression or CpG islands more like they control physical properties of the DNA, like relieving tension.
Thats why I am very interested in David Sinclairs above quoted research.
Have you read the paper yet, GaborB? Definitely interested in your take on it.
They showed that damage to retinal ganglion neurons in mice resulted in an increase in methylation age of those cells, and that in vivo treatment with AAV of OSK restored lost function. Interestingly for this discussion, they showed successful treatment by OSK was dependent on demethylases.
So, the likely sequence of events is that damage is inducing methylation changes, but that demethylation after the trauma can then restore function. As discussed elsewhere in this threat, the methylation is likely the after-effect of DNA damage repair, like a scar left after healing. Demethylation in my analogy would be akin to restoring the ability of skin to replace a scar with normal tissue.
They also mention that youthful methylation is not lost with this treatment, only that gained through age, or injury.
Hi Mark,
yes I have seen that paper – but in my post I referred to Robert’ interview with Sinclair where he had talked about unpublished results with more frequent DNA breaks accelerating aging.
(Although I believe accelerating aging is probably more easy to do and in many more ways than decelerating or reversing it)
I read the paper about the TET demethylases. Very clever paper. On one hand they are targeting an area where translation to the clinic may be the fastest. On the other hand they advance the general science a bit forward with the role of the demethylases.
What I am really waiting for is someone doing the in vivo OSK experiment with old transgenic c57 mice. Just to see the full potential of the treatment. I dont like experiments with progeric mice because we cannot be sure a gene defect that looks like aging is the same as normal aging.
GaborB – Presently, I am integrating small portions from it into my new film sequel “To Age or Not To Age -Transforming the human condition” – this film follows Guarante, Sinclair, Kenyon, Kennedy, Austad, Aubrey and others since 2006. I started it at Guarante’s lab in fall of 2006. I am making the raw interview available for Sinclair’s PR people
in the next week or so privately on Vimeo. At that point I will allow individuals to access that interview. In the interview, David is expansive, dodges no question,
no mater how personal and I even ask him if he anticipates attacks on his book.
Sinclair has been controversial in the past, but, having filmed him over a 12 year
period, I believe he is completely devoted and a true humanitarian.
Vow – if you could share the interview link with us we’d really appreciate it
Somehow my last comment was omitted. I am making the raw interview available to Sinclair’s PR people. I will make that footage available to individuals via private Vimeo soon.
@Robert
Please provide the link here.
Of interest to the discussion. I am sure Dr Alan Green will be happy to read this paper that Steve Horvath co-signed a few months ago: “Rapamycin retards epigenetic ageing of keratinocytes independently of its effects on replicative senescence”.
It is open access over at PubMed. This fits the findings so far that telomere length, proliferation or cellular medium (SC’s transplants) do not affect epigenetic age, although they correlate with it.
In fact, if taken at face value, those findings imply that some intracellular metabolic process is the main driver of the DMAm age signature.
The way I see it today, ageing is three pronged process: cell senescence and its inflammatory profile; loss of proliferative capacity and with it of organ regeneration; and change in gene expression profile leading leading to cellular dysfunction (think macrophages that are unable to clear SC’s or cancer cells; or osteocytes that are not able to maintain bone tissue). These 3 processes are related to each to other and I think the key lies in figuring out how exactly this is so, and how big a role each plays in ageing as compared to the others.
Yes, methylation is clearly linked to metabolism. I’m still waiting for someone to show how this causes issues.
Regarding your three prongs – they are also clearly related, as, for example cellular senescence and the inflammatory response are linked to loss of proliferative capacity through telomeres. Gene expression is likewise altered by telomere loss via TPE-OLD. Non proliferative tissues can also be induced to senescence through telomere damage via ROS and autophagy, and there is evidence in humans this happens in the heart. But probably quiescent tissues are mainly affected via the senescence of proliferative tissues, i.e. cardiomyocytes affected by the increased pumping demands caused by senescing endothelial cells. Similarly non proliferating neurons are affected by the aging of senescent glial cells.
That is why I still advocate addressing loss of telomeres as a solution to aging. I personally think this may also involve boosting progenitor cell numbers, and this may also cause a partial reversion of the epigenetic clock.
One would think that linking methylation changes to up and down-regulated genes would be easy. I am no researcher but I imagine the difficulty lies in determining the specific pathway and how in interacts with the cell’s behavior.
That’s similar to understanding how certain SNP’s correlate with a particular phenotype. Even for something as trivial and easy to measure as skin or eye colouring we have many SNP’s associated with specific traits but it’s a tall order to have a clear view of how it is that they work exactly.
Sometimes it’s very easy, as in a non-synonymous mutation yielding a non-working protein for a well known function. But think for example of the mTOR pathway and how complex it is and how many years it is taking to comprehend it well.
I echo your comments on telomeres. They offer a comprehensive theory of why we *might* age. We can build a logical argument about how they deplete tissues through senescence and change gene expression as they become shorter. But at some point we have to ask, why is it that they don’t correlate anywhere as well with age and mortality as the any of the epigenetic clocks?
Well we know inducing pluripotency reverses the epigenetic clock AND resets telomeres and mitochondrial function through various nuclear genes (HTERT, SHMT2, GCAT). The key is in connecting the methylation changes to those genes. assuming they are related at all. Or perhaps there is some intermediate ‘damage’ that causes the nuclear genes to change their function via methylation and inducing pluripotency removes the ‘damage’ and that causes the genes to reassert their proper function. For example it is possible to see how a cell would want to avoid excessive autophagy and mitochondrial biogenesis, as this process can be dangerous and lead to senescence once a cell has a certain backlog of aggregates it needs to clear. This is like the medecine that can cure or kill depending on the dose.
We’ve only seen mortality correlated with leukocyte telomere length so far. And leukocyte telomere lengths are notoriously unreliable. Even so, the correlation is linked mechanistically with aging. Something not yet done for the methylation clocks.
‘The epigenetic changes are what, at the nuclear level, triggers this dysfunctionality of the cell’, if these epigenetic changes are the same/similar as documented by the epigenetic clock, then these epigenetic changes /clock is accelerated when we are growing and by no means we can say that these epigenetic changes are causing us to grow old when we are young. in fact the body is overcoming these epigenetic changes and growing in spite of them.
we can’t say that these epigenetic changes/clock are the cause of aging. In fact it could be the cells with these epigenetic changes may be dysfunctional and reversing those changes could prove to harmful in the long run.
I think the CpG sites involved in the epigenetic clock are more like landmarks. Not necessarily a ‘counter’ in a loop programming analogy, the tick of a clock, not even a cumulative measure of dysfunction.
Think of it as the usual milestones in the life of the average person: first he’s born, then the baby shower, then goes to school, one day he graduates and so forth.
Going back to the programming analogy. If think these sites either store the ‘state’ of the cell, or are more like conditional flow statements. That’s probably how regular gene expression works to differentiate tissues: if cell is heart cell then express X genes.
Of course, are anything in biology is probably not so clear cut, and it contain a bit of every case.
Talking of youth, it is interesting to note that somatic cells that are immortalised in vitro continue to acquire these methylation changes. And yet they don’t seem to show any dysfunction. They continue to divide normally with a normal karyotype.
So in this case these methylation changes are not causing any harm.
I realise in vitro and in Vivo are completely different, but this seems to suggest to me that methylation changes are more of a counter of metabolic activity rather than a driver of dysfunction. At least in the case where telomeres are protected indefinitely.
Yes these epigenetic changes are not causing harm during the growth phase, indeed they may even be contributing to growth. therefore i suspect when you introduce young plasma in to an aged body, the epigenetic age/clock will not reverse, instead it will advance depending on the concentration of the growth factors.
during natural growth, the body has certain number of these epigenetically changed cells at peak youth. when we introduce young/infant blood plasma in aged body, the body will be having a larger population of these cells than at peak youth, the question is for true rejuvenation , the number of these cells have to match the number of cells at peak youth, otherwise fresh cells from growth factors signals will not have space to populate the organs.
Yes your suggestion that this could be a counter of metabolic activity or growth activity or this could be the aging clock. If growth and aging are two separate processes, then aging is always present, it is just that in presence of growth factors, the body produces more cells than the rate of the aging clock and when growth subsides, the normal aging clock takes over
Your suggestion of this being cell type dependent is quite interesting. For all we know, the methylation clock could just be measuring the type of cells present in the sample. For example I’ve suggested before that the line between stem, progenitor and somatic (and indeed senescent) cells is blurred. This clock might be reflecting that gradual change, both in the plasticity of the cells themselves, and in their relative numbers.
what I am hinting at is that the epigenetic clock shows aging starts when we are born, it could be aging is the natural state of the dividing cells/body after patterning/morphogenesis, but growth is another state imposed on the organism which operates in parallel. the rate of growth is far higher than the rate of aging (20x weight gain for the human male). Both growth and aging are occurring when we are young, but as growth tapers off, the natural state of aging continues.
what this means both aging i.e lifespan and growth i.e body size are selected in the ecosystem.
when we are growing, aging does not affect us because cells with epigenetic
changes who progress to senescence are effectively eliminated by the young immune system. But as growth stops , the it becomes increasingly difficult for the immune system to clear out senescent cells, which leads to age related dysfunction
Indeed. It may well be that the same signals that drive growth also drive aging, as suggested by Blagosklonny.
Growth signals prompt young cells to divide into more small, healthy cells and in parallel, accelerates the destruction of any impaired cells by cells specialised to that purpose .
Growth signals (even signals that are reduced by epigenetic regulation) prompt old cells that can no longer divide (or divide only sluggishly) to grow in size, and this results first in hyperfunction, and then dysfunction. Older, imparied cells are also tolerated and not destroyed, because the body knows it has no ready made replacements (better old than dead).
Hence stem and progenitor cells loss, telomere attrition and metabolic signalling are linked together in a network that drives growth, and then, aging. It just remains to see exactly how methylation patterns interact with this system.
Blagosklonny describes it as speeding car that can’t slow down. I think the car should be driving fast, that is youth. It is just that the car and road system (to stretch the anaology) needs to be maintained by proliferation competent cells. But humans are only designed with a set about of cell divisions. Hence aging and death is inevitable.
But why does the body tolerate these epigenetically changed cells, when it can easily eliminate them even when it is in a growth phase and it can easily replace these cells. But the body does not do it even when it is growing strongly as evidenced by the epigenetic clock. I would say a subset of genes from both the growth and aging processes are involved in the initial patterning/morphogenesis and when that process is complete, twin processes of growth and aging are activated with turnover rates and therefore both these processes cannot be selected against
What do you mean they cannot be selected against?
I do know that unlike DNA damage where there is a template to effect repairs (the other strand for single strand breaks and the other identical chromosome for double break damage), there is no epigenetic template to tell a cell it has gone astray. The only template would be the epigenetic state of the progenitor parent cell a given cell line is sourced from, and if there is nothing overtly wrong with the cell in question (so it is not destroyed by other mechanisms such as cellular senescence or apoptosis), then the cell line will continue in its altered epigenetic form until such time as it is dysfunctional enough to be recognised and destroyed. This mainly applies to epigenetic ‘noise’. Intentional epigenetic changes will have been made according to other problems (such as having no ready made replacements), and therefore will not be corrected unless the other problem is first addressed.
I mean the subset of genes that are involved in aging and growth may also be involved in early embrogenesis/ patterning.
To test the function of methylation changes during growth, these modified cells can be eliminated in young mouse models to see
1- growth is retarded due to their elimination
2- their elimination leads to uncontrolled growth and vice versa their presence indicates that their increasing population leads to growth maturity
3- growth is normal even after their elimination, but lifespan is increased
I see.
You may find there is no easy way of removing them until they become dysfunctional in some way, and hence express some easily targeted marker. And if they are not yet dysfunctional, this explains why the body would not remove them and then have to replace them using valuable, limited cell divisions. According to this argument, removing them (if possible) would decrease lifespan.
Thanks for the valuable discussion.
But companies doing partial reprogramming are showing that these cells are dysfunctional and the reprogramming protocol makes them young again
What i am trying to say , there has to be a way to isolate their function during the growth phase, and if we find that eliminating them during the growth phase has no deleterious effect, then we can maybe say that they are there only to implement the aging program
Not exactly.
Reprogramming rejuvenates cells AND resets methylation profile of old cells. We do not know that resetting the methylation profile then leads to rejuvenation. It may be the two effects are only correlated due to something else that you are fixing through reprogramming, for example telomeres and mitochondrial function.
You are right we do not know resetting the methylation profile leads to rejuvenation in the true sense, and whether those cells are identical to their young counterparts and the effect of rejuvenation may also effect something else as you say telomeres and mitochondrial function, which could also be detrimental.
The argument is that evolution is stuck with a good effect and a bad effect of the same genetic substance, and evolution has had to compromise, and aging is the result.
It’s a really old idea, going back to George Williams in 1957, and it has currency because it provides a way for old-school evolutionary biologists to avoid moving ahead to consider diverse modes of natural selection, beyond the “selfish gene”.
Evolution has shown great ingenuity in separating two antagonistic effects in other circumstances. It is commonplace for a gene to be duplicated in the genome (it’s called a “transposon”), and then each copy goes its separate ways. It’s even more common for genes that are beneficial at one time of life to be turned on then and only then, turned off forever after. Thousands of genes are active only in early stages of development, and they are efficiently silenced for the rest of life.
So I’m skeptical about the argument that evolution was caught between a rock and a hard place, and had no other choice but to accept late aging as a price to pay for early growth.
There’s a whole chapter on this in my book.
i believe evolution in an iterative manner arrives at an optimal organism for a particular ecosystem, wherein both the organism and ecosystem survive. In that scenario ecosystem will evolve an optimal path, and that path may involve a good and a bad effect of the same gene. It may not be a compromise but just an optimal solution.
On a side note, i would say this is the best ever blog on aging with really stimulating articles and comments.
thank you josh
Is late aging really late aging or normal aging which is always there and starts after we are born, but which is camouflaged by exponential growth when we are young.
are growth and aging on a continuum or two separate processes with vastly different cell turnover rates. It is a question that the methylation clock throws up
I think the confusion is also semantic. The epigenetic clock tracks ‘age’, so it certainly is picking up morphogenesis and developmental signals in early age. And if you regard ‘old age’ as a continuation of development then it is tracking the same thing at those later stages too.
If you shed the idea that ‘ageing’ is the accumulation of damage, whether due to growth going awry or just random irreparable damage, then it all fits.
“Old age” would be the developmental epigenetic program manifesting itself, just like it drives growth and maturity in your childhood and teens.
Take for example grey hairs in humans or gorillas. Is that ‘damage’? or is it a sign of maturity? Is it a ‘mistake’, an unavoidable error, or a ‘goal’ programmed in our genome?
Regarding methylation being correlated or causal in aging, having read Sinclair’s latest work, I have to conclude that it is mechanistically involved. Damage (such as to retinal cells in Sinclair’s work) results in methylation (like a scar being left after (DNA) healing), and treatment with OSK results in the resolution of that damage. Most importantly for our discussion, the success of OSK treatment is DEPENDENT on demethylases.
My guess is that is you demethylate enough, you’ll activate genes responsible for a more stem-like state, with more regenerative and proliferative (telomerase) ability.
Perhaps this is part of the development program as you suggest, and it’s related to the set body size we obtain. It’s interesting to note that in Sinclair’s work, demethylation does not affect methylation patterns set down in youth, only those gained later. So presumably this treatment would not accidentally make you a child again.
It would be interesting to know what partial reprogramming does to epigenetically young cells in vivo as opposed yo old cells, the publications on partial reprogramming do not address this
Sinclair’s work suggests youthful epigenetic marks are not removed, only those acquired by aging or trauma.
Most likely the continual proliferative ability of immortalised cells in vitro overcomes any harm from accumulated changes in methylation. This is because the cells stay small through division and do not accumulate metabolic waste. My guess is that this aging effect only kicks in at the cessation of growth/ start of adulthood.
It might be instructive to examine methylation changes in the circular DNA of bacteria. That could shed some light on this. Anyone know of any studies that have been done?
“Most likely the continual proliferative ability of immortalised cells in vitro overcomes any harm from accumulated changes in methylation”.
Those cells stay alive because a petri dish is not a highly organized multi-cellular animal. The epigenetic marks we are discussing don’t necessary harm or kill a cell. They kill the ‘host’, eventually, by ‘poisoning’ the environment or failing to repair it as they were once used to doing.
I don’t think the DNAm clock is a compensation mechanism, or a passive time keeper like rings on a tree. I think it is an active developmental process from conception to death.
On the contrary, cells in a petri dish are not ‘maintained’- typically they have much stronger growth factors, higher O2, higher ROS, lose telomeres more quickly, differentiate faster and accumulate epigenetic methylation changes at a much greater rate than the tissues in a body (the last as shown by Horvath).
Of course, the body IS far more complex than a petri dish, with many different types of cell supporting one another. For example, we can look at neurons, which don’t divide, but which do accumulate metabolic waste. The rate they accumulate this waste is dependent on the assistance they have from glial cells. Glial cells senesce through telomere attrition. Hence, we have neuron cells accumulating waste and damage (in the SENS sense of the word), adapting epigenetically (as well as randomly) according to the Horvath/Sinclair model, but this is somewhat dependent of telomeric aging in other cells. Probably the single best point of intervention would be telomerase, though for some long-lived waste perhaps SENS will also be important. Of course, once neurons are lost, or seriously damaged, no amount of telomerase will bring them back – we need stem cells, and for this epigenetic reprogramming or an exogenous supply will be required.
I think procaryotes dont methylate their DNA. They dont do sex dont do elaborate body plans and dont age. I think those things are correlated. They dont have telomeres either.
Yes – mostly true. But some bacteria have found a way to age, paradoxical as that seems.
https://www.ncbi.nlm.nih.gov/pubmed/20735346
Does that mean we don’t methylate our mitochondrial DNA?
This more recent paper is informative Josh – and shows asymmetrical division of damage under stressful circumstance is the method by which bacterial lineages allow some lines to survive.
https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.3000266
If the clock is tracking age, rather being the cause itself, partial reprogramming would not result in rejuvenation.
If partial reprogramming is resulting in rejuvenation, by erasing the methylation marks, it can be said that the methylated cells are dysfunctional. If these cells are dysfunctional, why does the body not eliminate them, rather it accommodates them from the time we are born, this leads to the question that whether these cells give rise to the aging program
The research so far doesn’t point to those cells being dysfunctional. Rather, ALL cells in a body would tick a similar rate over time, setting off stages in growth and development. It is only at the later stages in life that those marks would turn those cells dysfunctional. Presumably, if the evolutionary aging theory is right, on purpose.
Nobody knows for sure we are looking at an aging clock, or at the only one. Perhaps simple senescence cell accumulation is the main driver and it is a somewhat independent process (likely, it isn’t, and is is connected to epigenetically aged immune cells). Perhaps, turning back the clock does not repair much damage, just stops it.
But it had been speculated for long that aging was an epigenetically driven process. After all, the germ line is immortal, and bar random DNA mutations, we have the same DNA at birth and at death. Children inherit the mutated DNA from the parents, and they are not born ‘old’. So what has changed? Gene expression seems like the most likely driver.
partial reprogramming is strongly hinting, if not proving that these cells are dysfunctional in an aged body. the only way to know whether these cells contribute in anyway during the growth phase is to eliminate them during those phase and isolate their function.
when it is shown that these cells do not contribute to the body in the growth phase, we can then maybe speculate that these cells are part of an aging process
I think maybe we need to be more nuanced in our use of the word ‘dysfunctional’. When I damaged my knee doing Judo, the ligament healed thicker but shorter than before. This made it more resistant to a similar injury in the future, but impaired my range of motion. You could say that my joint ‘got old’. But it was a kind of adaptation. Perhaps cells are similar – epigenetic changes could be a memory of past insults. This would decrease their plasticity and adaptability but specialise them to being resistant to the past threat. Rather like your Grandad who still harbours a grudge against the Germans from WW2, even though we’re friends now.
Maybe these adaptations are beneficial up to a point, but eventually become burdensome to the body.
Such good scientific reasoning, but it turns out that’s not the way nature has decided to do things. Surprisingly, that effect has little to do with aging.
I was reading a paper on the use of viruses to promote an immune response against cancer cells, as well as to directly attack such cells and
/or to deliver toxins to the cells. I recalled Josh’s interesting essay and wondered if anyone is researching the use of viruses against senescent cells?
in the post in march 2019 ‘DNAm GrimAge—the Newest Methylation Clock’ by josh, a study was mentioned ‘Blood stem cells from the bone marrow were transplanted for medical reasons, and years later, the blood cells derived from the donor stem cells were collected and analyzed for methylation age. The result was that the blood cells remembered the age of the donor’.
the study in Aging cell also mentions ‘The present study shows that the DNAm age of donor blood is not influenced by the environment of the recipient’s body, whether younger or older, and that the DNAm age continues to increase after transfer to the recipient’s body as if the donor cells were still in the donor’s body. This trait persisted even 17 years after the transfer to recipients who were 1 and 3 years old at the time of HSCT’
In this study, another conclusion can also be drawn besides the one that is implied i.e donor blood form aged persons infused in recipients who are 1 and 3 years old, with a higher Dnam age/higher number of methylated cells
does not influence the normal growth of the recipients.
Are the studies results really that suprising? The donor HSCs are at the top of the tree, supplying all progenitor and somatic cells that they give rise to. Therefore you’d expect the donor to influence methylation age of the recipient’s blood, but not the other way around.
It also suggests methylation age might not the be-all-end-all of biomarkers when it comes to health and aging.
the study also hints that the donor cells which have a higher methylation age do not hinder growth or dysregulate growth in infants, the recipient body accommodates these additional methylated blood cells with a donor dnam age
That’s a very good point Kunal. But in terms of development even a 1 year old baby is very ‘developed’. Morphologically he is the same as an adult, only needs to grow and the tissues to mature/strengthen.
I think doctors/researchers are already looking into the progress of transplants depending on the age of the donor. I think S. Horvath made some comments on this in some of his talks, but I would need to re-watch them.
In theory it is not a matter of these cells ‘accumulating’. But all of them ticking away with a predictable pattern of methylation, even if there are differences between tissues.
this study just shows transplant of aged blood stem cells in infants, but still why would the body expend energy in maintaining them in higher numbers as the study shows and not eliminate them, when they do not contribute to growth and eventually contribute to increasing senescence in the aged body.
On your point that ‘even a 1 year old baby is very ‘developed’. Morphologically he is the same as an adult, only needs to grow and the tissues to mature/strengthen’
i would argue the four major approaches of the researchers in tackling aging depend on the point you have made.
1 young blood plasma growth factors
2 partial reprogramming
3 stem cells
4 eliminating senescent cells
all the above approaches assume that all the organs in the body have enough information to regenerate and regain peak function, if dysfunctional cells are removed and growth factors are made available, because if a one year old baby can do it who is 1/20 the weight of an adult, then the aged body can surely do it.
Young blood cocktail stops Alzheimer’s decline, early clinical trial reports
https://newatlas.com/alkahest-young-blood-plasma-alzheimers-cognitive-decline/60927/?fbclid=IwAR3_Tk3HS9ovTzvQlELXztsb1FcRdAtZcJDl9Jxhhb2D7wRE_0g7uYYUn_E
I believe that it does work in animals and humans.
Here is my case:
Looking into how Turn Bio is reversing the cells aging.
The article
An Interview with Prof. Vittorio Sebastiano of Turn.Bio
says that
“You are using Oct4, Sox2, Klf4, and c-Myc (OSKM) but also LIN28 and Nanog to make OSKMLN”
I am going with Oct4 is it appears to be critical to the process.
So how do we turn on OCT4?
The study:
Tryptophan derivatives regulate the transcription of Oct4 in stem-like cancer cells
states:
“Reduction of endogenous ITE levels in cancer cells by tryptophan deprivation or hypoxia leads to Oct4 elevation.”
So now we have 2 ways to turn on OCT4
Low tryptophan
or
Low Oxygen levels.
So the question is, does low oxygen levels in nature promote longevity?
The article:
Naked mole rats can survive 18 minutes without oxygen. Here’s how they do it
states
“The animals can survive more than 18 minutes without oxygen.”
Article:
Dead zones enhance key fisheries species by providing predation refuge.
states
“only the harvested quahog clam (Mercenaria mercenaria) thrived in hypoxic areas”
Both the quahog clam(500+ years) and the naked mole rat (20+ years) live in low oxygen conditions.
So what about us humans?
(the following is for information only, I am not promoting either)
The article:
Effect of moderate alcohol intake on nocturnal sleep respiratory parameters in healthy middle-aged men
states:
“drinking alcohol before retiring resulted in lower arterial blood oxygen saturation (SpO2) during the early half of sleep”
also
The article:
Cigarette smoking decreases tissue oxygen.
states:
“smoker experiences tissue hypoxia during a significant portion of each day.”
So what I would expect is that we would see that smoking and/or moderate
drinking associated with longer life.
In your recent article on the new clocks noted that heavy smoking is better than
2nd hand smoke for longevity.
This might be explained by the increase in Oct4 levels(low oxygen).
We have all heard limited alcohol drinking seems to have health benifits.
and
in various studies of the 100+ crowd, that many smoke(low oxygen), drink(low oxygen) or are vegitarians(low Tryptophan)
The following is just my personal experience.
My wifes great great Grandma lived to 104 years, while her 2 sister only lived
into there 70s.
We know that before bed each night she drank a glass of sherry(low Oxygen).
We visited her just before she passed away, looking at her I would not have
thought her more that 50 or 60 years old.
Her own doctor remarked while she was in her 90s that she had the heart of a 50 year old.
this is pure speculation
suppose aging is a separate process from growth, then there could be a separate growth clock because the Dnam clock is looking at complete lifespan, and the growth clock stops at maturity.
but then how does growth stop if aging is a separate process, another speculation, there could be growth maturity clock which maybe starts at puberty.
I repeat this is pure speculation
About the study in Aging cell, an observation
From the study
DNAm age of donor blood is not influenced by the environment of the recipient’s body even if the recipient is 1 year old, this trait persisted even 17 years after the transfer to the recipient and the DNAm age continues to increase after transfer to the recipient’s body as if the donor cells were still in the donor’s body and effectively the blood cells remembered the age of the donor.
suppose the 1 year old recipient received blood from a 50 year old recipient, and after 17 years, the recipient’s blood age is roughly 68 yrs.
The Dnam clock shows that epigenetic age rises exponentially when we are growing, but in the case of the transplant to a 1 year old, the dnam cock advances at a rate, as when the body has already matured.
So is advancing at an exponential rate just an illusion due to a very high rate of growth or is it advancing at the same rate, as when the body has already matured.
what effectively is happening
neither the Dnam age has any effect on growth of the blood cells in the recipient,
nor the growth of the recipient has any effect on the Dnam age of the donated blood cells
Google “MicroRNA“ – and you will see that the aging program is implemented through MicroRNA.
More information would be helpful. This doesn’t give us much to go on.
This study https://www.nature.com/articles/s41598-019-39919-3
‘Synchrony and asynchrony between an epigenetic clock and developmental timing’
it is about the relationship between the epigenetic and the developmental clock and a snippet from the study ‘Taken together, these results suggest that even though the DNAm aging clock and the developmental stage typically correlate with the gestational age, these clocks can be decoupled in some cases and likely represent two independent timing mechanisms.’
That’s a very interesting study Kunal. Thank you for sharing.
I was recently reviewing some of S. Horvath talks and he mentioned he believed his clock was tracking development. This study would imply that the processes are somewhat separable.
I suspect there is a bit of both, as it is likely that many of the sites chosen by the algorithm also track development and growth by association. As Gabor commented above, there are many possible sites that can be selected, but one thing to keep in mind is that the original Horvath clock is multi-tissue, so many of the signs of cell differentiation would not be selected.
One possible interpretation of the studies so far is that the clock is driven by metabolism and proliferation. As cells multiply and function a set of sites vary their methylation as they progress from a young state to an equivalent old one. This process starts from conception and throughout life and results in a particular average methlylation state of the tissue. The higher the proliferation and metabolic rate the faster it ticks. So all tissues would have some percentage of old and young-methlylated cells.
Another take is that the process is more tightly controlled across the same tissue and most cells have the same methylation state correlated to their proliferation and metabolic rate history.
The way I understand the DNAm clock to work it would not differentiate between the two possibilities. In practical biological terms, they may not make a bit difference either. Both resulting in predictable outcomes as a result of average gene expression.
There’s also the question of whether individual sites are more likely to vary at a particular point in the aging of the cell, which would correlate more with a controlled ‘ageing’ mechanism. The studies that utilize one or very few sites are clocks seem to imply this.
A snippet from the study
‘ comparisons of the epigenetic clock from different regions of the retina revealed no major differences; different parts of the retina followed the same trend as the whole retina and maintained their epigenetic age, even though central retina is developmentally accelerated compared to peripheral retinal regions . This result suggests that the developmental clock and the epigenetic clock might be controlled independently.
Maybe that is why young blood plasma rejuvenates the aged body, it basically rejuvenates the cell without reversing the aging clock using a different growth mechanism
This study is about Link Between Epigenetic Clocks for Aging and Senescence.
‘On epigenetic level, replicative senescence and aging evoke characteristic modifications in the DNA methylation (DNAm) pattern, but at different sites in the genome’
https://www.frontiersin.org/articles/10.3389/fgene.2019.00303/full
This study ‘Epigenetic ageing is distinct from senescence-mediated ageing and is not prevented by telomerase expression’
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6224244/
Let us for a moment assume the aging mechanism and the growth mechanism are separate circuits in the cell, then is it possible that these mechanisms are targeting common genes through histone modification in an antagonistic fashion, with the growth mechanism having precedence. The cell divides and functions optimally during the growth phase, but as the growth impulse is suppressed due to a maturity milestone having reached, the aging mechanism starts to dominate and progressively makes the cell dysfunctional.
This is the explanation I wrote for epigenetic aging back in March 2018 on one of Josh’s other posts. It still seems true to me.
https://joshmitteldorf.scienceblog.com/2018/03/19/telomerase-update-and-downgrade/#comment-391343
What if epigenetic age is just a measure of time since a somatic cell line was spawned from it’s stem cell progenitor (which by definition will be younger epigenetically)? That means older people would naturally have an older epigenetic age because their stem cells are less active and somatic cells are replaced less often. It also means an individual with sightly more telomerase (and longer telomeres) would naturally have longer lasting somatic cells, so at any given time their tissues would be older epigenetically because they are also replaced less often. If this is true it means there are two separate but easily confused correlations: older people having less often replaced somatic cells due to tired stem cells, and people who have longer telomeres needing replacement less often (but probably with better preserved stem cells as a result).
I have a perspective based on genetics and evolutionary history that makes me dubious that this is what’s going on.
There’s also the fact that Horvath has designed a “multi-tissue methylation clock”, that gives the same reading for blood cells (very recently spawned) and kidney cells (much older) and heart cells (probably as old as the body itself).
But I’ll ask you: suppose we were to design an experiment to test whether what you say is true. What would the experiment look like?
The first part of the experiment is simple – see if telomerase activators cause an increase in epigenetic methylation age in leukocytes (or buccal cells, for example). Even if the telomerase activator is only moderately effective, it should reduce how often the somatic line is replenished from the underlying stem cell niche.
The actual chronological age of somatic cells (blood vs kidney for example), may not matter – as it is likely the underlying progenitor cell pool (the stage between stem and final somatic cells) that ages.
I would go with steve horvath’s hypothesis in his 2013 paper that this is an epigenetic maintenance system
I have found this interesting paper from 2013
Proliferation Rate of Somatic Cells Affects Reprogramming
Efficiency*
THE JOURNAL OF BIOLOGICAL CHEMISTRY
They claim that OSK is more effective in IPSC generation then OSKM,, and cMyc is only needed to turbocharge already transforming IPSC colonies.
If this largely sidelined research is true, then cMyc a known oncogene should not be used in in vivo reprogramming experiments thus the treatment window might be widened or the dose increased without risk of malignant transformation.
I have come to this paper by speculating that instead of the slow mouse experiments, researchers should use serum starved confluent monolayers for rejuvenation studies. They could grow the primary cell culture from old donor cells, then stop serum and start interval OSK treatment followed by functional assays (like profiferative capacity, beta galactosidase, lipofuscin, telomeres, mDNA age etc) .
That could save time and money.
This is all interesting work. But in my opinion we can relax – in Vivo reprogramming with all its risks simply is not required. IPSCs can be made from any somatic cells, expanded in culture (telomerase already present) and injected via IV to the patient. No terratomas are formed this way and the tiny cells can go anywhere in the body they are required to effect repairs.
https://www.ncbi.nlm.nih.gov/m/pubmed/30662568/
So looks like we have a real rejuvenation therapy on our hands. Anyone know a good stem cell clinic that will do it?
well I am not convinced based on the scarce research available at this timepoint that interval OSKM really works in wild type organisms and really is anything more than turbocharging a wound healing mechanism.
also I have some doubts about injecting manufactured solid tissue cells. from what I have read these cells usually end up in the lungs and die in a few days..
so I think we should understand the biology of rejuvenation and then go for small molecules approach
or the ex vivo culturing of stem/progenitor cells you mentioned coupled with senolytics and blocking scar formaton this one could be a better solution but I dont see it coming in the next 20 years
Why would it not come in the next 20 years? Both senolytics and iPSCs via IV are available now! I don’t believe that the problems with MSCs occur with iPSCs due to their much smaller size and pluripotency. Also, they don’t last long in the body because the body’s own signalling causes them to quickly differentiate as required (which is what they want). IPSCs are the best cell to add to the human body because we don’t have any (after the embryo-fetal transition) and they are right at the top of the chain, renewing and refreshing all cells beneath them. It is the perfect combination with senolytics. Better even that telomerase therapy, which can potentially keep some cells that would be better off replaced, alive.
IV can’t really be done with small animals though, so this is a straight to human trial.
Would be great but is there any evidence for IPSC engraftment in vivo?
From the abstract of the paper I posted
‘Intravenously administered iPSCs were therapeutic with a dose as low as 5×10^6/kg and some iPSCs differentiated into somatic cells in injured organs. Disseminated iPSCs trafficked into injured tissue and survived significantly longer in injured than uninjured organs. In disease-free animals, no intravenously administered cell differentiated into an unwanted long-lasting cell or survived as a quiescent stem cell.’
Very promising indeed.
yes, promising, but I miss a few things: engraftment into normal tissue – they reported the opposite of this
biomarker improvement – e.g. in case of kidney injury improvement in creatinine clearance to controls
lifespan improvement
all they proved IMHO that iPSC can safely integrate into site of damage, but what if it is only because iPSCs can form scars much more effectively.
we have to resuscitate dead nephrons, dying neurons and glia and the like to achieve life span extension
That wasn’t my interpretation of the results at all – but I will re-read the paper to be sure of my facts.
It seems highly unlikely that iPSCs would form scars – this is what less plastic cells do in the absence of the requisite stem cells. The whole point of iPSCs is that the signalling of the environment they find themselves in decides what they differentiate into (and only form terratomas if their own signalling becomes dominant – which is easily prevented if they are diffuse.)
In theory, with the right signalling iPSCs can replace any type of cell, including neurons. This obviates the need to reprogram old cells back to health – simply allow them to be gradually replaced.
Even more excitingly, iPSCs could with the right signals regrow limbs and give humans fully regenerative abilities.
Where has your sudden pessimism come from Gabor?
well, I am not saying that iPSCs only contribute to scars, what I say is there is no proof yet that iPSC IV can be used for rejuvenation. also the evidence on interval OSK(M) is not very abdundant at the moment
my pessimism is mostly about the sluggish progress in the aging research and not entirely new. I think eventually cell replacement will be used, but currently I see this 50 years away – similar to fusion energy and the likes
Well this paper only came out in Jan 2019, let’s hope others take up the baton.
I agree that the pace of research into aging is way too slow. It should be treated as a Manhattan Project or Apollo Moonshot, but sadly not all people think as we do.
Small study on reversing epigenetic aging:
https://www.nature.com/articles/d41586-019-02638-w
I cannot find the original article in Aging Cell.
If somebody can, please drop a link.
Looks like a case of growth hormone waking up quiescent stem cells to me. Which may not be a good thing in the long run. Assuming I’m right about what epigenetic age is basically measuring.