

I have been enthusiastic about telomerase therapies for anti-aging since 2003. But if I can’t change my mind as new data appears, what’s the point of being a scientist? I still believe that lengthening telomeres is a net benefit, but the potential for added years is modest, and there are probably risks and tradeoffs. The study that has most influenced me is this one, implying that telomerase affects epigenetics (through methylation) in ways that accelerate aging. My theory is that the unexpected relationship between telomerase and methylation is an example of antagonistic pleiotropy, but pleiotropy in a very different sense from the standard evolutionary theory.
Do people with longer telomeres have longer life expectancy? In 2003, Richard Cawthon of University of Utah first addressed this question experimentally with a study that was clever, innovative and courageous. It was innovative in that he introduced a fast and convenient way to measure telomere length from very small quantities of DNA, using the Polymerase Chain Reaction. It was clever in that, instead of a “prospective study” measuring telomere length in his subjects and then following 20 years to see what would happen to them, he did the experiment retrospectively, using historic samples of blood that had been taken from people twenty years earlier and kept in frozen storage by a local hospital. And it was courageous in that everyone believed at the time that extending life could not be so easy as just lengthening telomeres, or else the body would already be doing it! That is to say, no one would fund the study because they thought they knew how it had to come out.
 But they were wrong. Even with Cawthon’s small sample of only 143 subjects, the relationship between telomere length and diseases of old age jumped out of the statistics. The quartile with the shortest telomeres had suffered two times higher mortality and three times greater incidence of heart disease in the intervening 20 years than those with the longest telomeres.
But they were wrong. Even with Cawthon’s small sample of only 143 subjects, the relationship between telomere length and diseases of old age jumped out of the statistics. The quartile with the shortest telomeres had suffered two times higher mortality and three times greater incidence of heart disease in the intervening 20 years than those with the longest telomeres.
Red blood cells have no DNA, hence no telomeres, but white blood cells are constantly dividing to target specific bacterial, so the telomeres in white blood cells are a sensitive measure of immune health. Cawthon reported that the group with shortest telomeres had suffered 9 times the rate of infectious disease compared to the longest telomere group.
At the time of Cawthon’s study, there was a great deal of skepticism, based purely on theory. The standard hypothesis was that all animals are evolved to live as long as possible, all else being equal, and if telomerase were being held back, there must be a powerful downside associated with it. I was already marching to the beat of a different drummer in 2003, and I didn’t believe that evolution was always going for the longest lifespan available. Because I believe that aging is an evolutionary program, it was easy for me to see telomere shortening as part of the program. The biggest clue in my mind was the evolutionary origin of telomere shortening in single-celled protozoans. In the ciliates (e.g. paramecium), telomerase is not expressed in mitosis (when the cell copies itself), but only when it conjugates (recombining genes with other individuals) with another. Hence, a cell that just goes on reproducing as fast as possible without sharing its genes was doomed to die of cell senescence. A billion years ago, telomeres were already a means of enforcing the communal imperative, Share your genes! It is easy to imagine that the same evolutionary imperative has persisted through the aeons, and that telomere shortening insures death in many higher organisms. Indeed, since Cawthon, it has been demonstrated that short telomeres are a mode of aging in dogs, cats, and horses, (but not cows, pigs or mice).
Three years ago, I reported on a Danish study that replicated Cawthon’s results on a huge scale. In 60,000 subjects, Rode associated short telomeres with all-cause mortality, heart disease, diabetes, and some cancers.
Telomere shortening leads to senescence and higher disease risk by three known mechanisms. First, stem cells with the shortest telomeres stop reproducing, hence the body’s tissues don’t renew as efficiently. Second, senescent cells are not just dead weight, they actually emit chemical signals (cytokines) that increase inflammation. This has been called SASP, for Senescence-Associated Secretory Phenotype.) Third, senescence in the bone marrow that generates new white blood cells is especially damaging to the immune system, because it prevents the body from responding effectively when challenged with new infections.
Telomeres and cancer
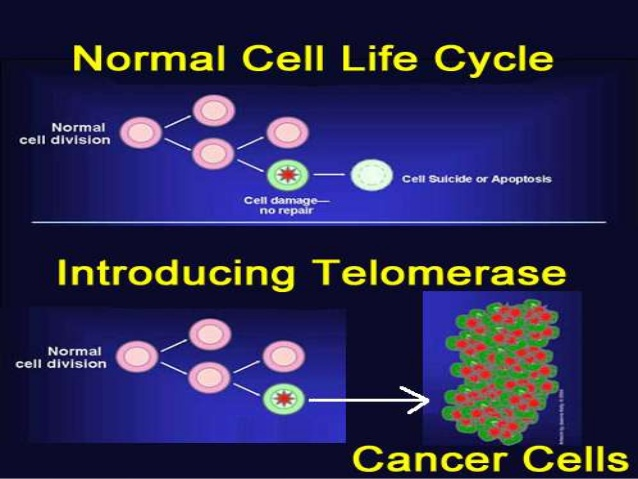
If short telomeres cause all these problems, why would the body ever allow its telomeres to become short? It was recognized early in the game that production of telomerase entails no substantial metabolic cost, so the question challenges the conventional theory that individual animals are evolved to live as long as possible. Of course, for us who believe that aging is programmed, there is no problem with this. But the first suggestion of an answer within the conventional paradigm came from Carol Greider, one of the original discoverers of telomerase, and independently from Ruth Sager. Telomerase is needed to make cells immortal. 90% of cancer cells have found ways to bypass the suppression of telomerase in order to continue proliferating unabated. Greider and Sager proposed that keeping telomerase under lock and key constitutes one of the barriers that keeps cells from going rogue as tumors. Telomere shortening helps to prevent cancer.
This argument never made any sense to me. First, what good was it to suppress cancer if the net effect was to shorten lifespan? And second, I believe that the body’s principal defense against cancer is the immune system, and if short telomeres can cripple the immune system, that was likely to do more to promote cancer than to prevent it.
Nevertheless, the idea that telomerase is rationed to protect against cancer persisted in the biomedical community for 20 years based on theory alone, even as it was moderated by the discovery of SASP.
Experimental link between telomeres and cancer
I came into this field very skeptical of the idea that long telomeres could cause cancer. But as the evidence has accumulated, I’m compelled to reconsider. Just last summer, I blogged critically about the largest genetic study to date, linking genetic predisposition for longer telomeres with cancer rates later in life. I noted that the measured effect is actually quite small, but is reported blown up to alarming proportions by exponential extrapolation. But that didn’t mean it was necessarily wrong, only that it was unconvincing. Shortly afterward, I became aware of observational studies, based on measured telomere length rather than the genetic predisposition. These are harder to refute.
In this study from the Moffitt Cancer Center, short telomeres (as measured directly, not imputed from genetic variation) are associated with higher risk of squamous cell skin cancer, but long telomeres are associated with higher risk of melanoma skin cancer. Same methodology, same authors. Why would I believe one and disbelieve the other? Melanoma tends to occur at younger ages than squamous cell carcinoma, this supporting the Greider hypothesis that telomere shortening should be especially important for cancer prevention while we are still in a fertile stage of life. The Moffitt results on melanoma were confirming a finding reported earlier from Harvard Med School.
In this study, people with the longest telomeres had nearly twice the risk of lung cancer compared to people with short telomeres, after adjustment for age and smoking status. There are 25 co-authors, and Cawthon is #2. In this study, short telomeres protect against (devastatingly lethal) pancreatic cancer, and in this one, there is an elevated risk of breast cancer associated with long telomeres.
There are studies contradicting each of these findings. Overall, the field seems to be more of a confused mess even than most areas of epidemiology. But for lung cancer, melanoma, and pancreatic cancer, the predominance of the evidence says that longer telomeres are associated with higher risk.
Longer telomeres uncontroversially protect against heart disease and stroke. There is no contradiction of this finding in sight, and there has been no contradiction of the major finding (by Rode and Cawthon) that short telomeres increase all-cause mortality. Perhaps that’s all we need to know.
Stop the Presses
Just a few weeks ago, I learned of this new study linking telomerase to the epigenetic changes that the methylation clock associates with aging. The implication is that telomerase accelerates aging. It began with an investigation by Steve Horvath’s group (about which I reported last month) asking, what genetic variations are associated with people who age faster or slower than average, according to the Horvath methylation clock? They did a genome-wide search for statistical correlates and the standout association was telomerase. People who have small genetic variations that support greater telomerase expression tend to have longer telomeres, but they also tend to age faster, as measured by the Horvath clock.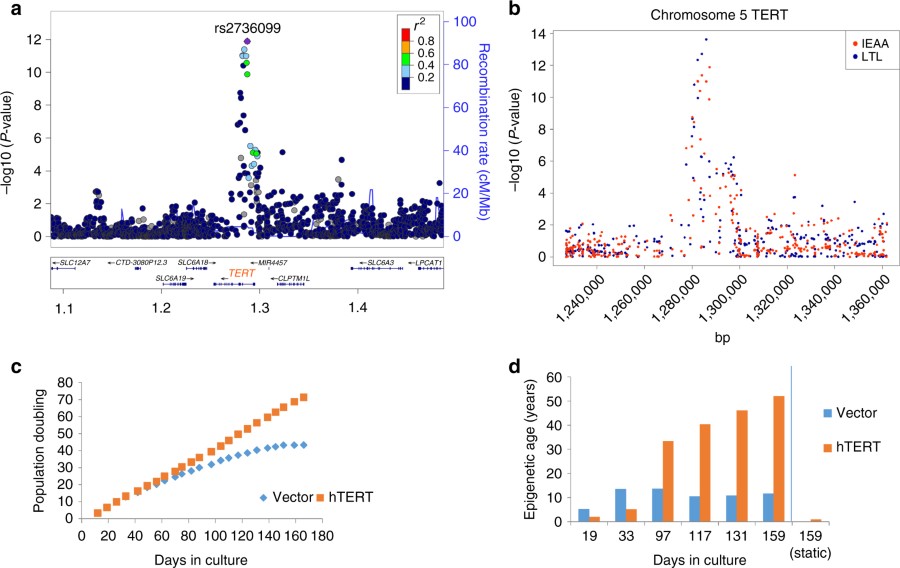
It’s been known for a long time that telomerase has other effects in addition to lengthening telomeres. But this is the first time that telomerase has been reported to affect DNA methylation. So it seems we are presented with a tradeoff, or pleiotropy, or Catch-22, or “damned if you do, damned if you don’t.”
The association between telomerase and accelerated aging (measured by methylation) was found in the genetic statistics, and then confirmed in a cell culture. When telomerase was artificially activated in the cell culture, the methylation patterns changed in the cells consistent with older age according to the Horvath clock. In fact (and remarkably in my opinion) they found no Horvath aging at all in the cell cultures that lacked telomerase. Could it be that telomerase is the one and only driver of epigenetic aging at the cellular level?
Telomere length and the Horvath methylation clock are both correlated with age, but they are not otherwise correlated with each other. The Horvath clock is a combination of 353 methylation levels that is optimized to correlate maximally with age. The observed correlation is 0.95. Telomere length is not statistically optimized but measured as nature offers it, and its correlation is much weaker (~0.4 according to my estimate, as I have not found this number in print). Thus Horvath clock is an excellent measure of chronological age, and combining information about telomere length can make it potentially a little more accurate yet. But the telomere clock on its own is a very unreliable measure of age.
The Horvath group designed an experiment to separate the direct effect of telomerase on methylation from an indirect effect (telomerase ⇒ telomere length ⇒ methylation age). They found no indirect effect. Telomerase itself affects methylation aging, but telomere length does not.
This raises (what is for me) an uncomfortable question. Many “good” life habits have been associated with telomerase expression, including exercise, meditation, and social integration. Could it be that these habits are simultaneously slowing our telomere aging, while hastening our epigenetic aging?
“While the paradoxical finding cannot be disputed on scientific grounds, its biological interpretation remains to be elucidated.” [Lu et. al, 2018, the same study I’ve been talking about]
(Another finding of this same study: Earlier menopause is associated with epigenetic age acceleration in women, but this is mitigated by hormone replacement therapy. HRT modestly slows aging, as measured by the Horvath clock.)
Antagonistic Pleiotropy turned Upside Down
So, what’s going on? My inclination is always to think in evolutionary terms.
Antagonistic Pleiotropy is the standard explanation for aging, though I have long argued that it doesn’t fit the data. The theory says that some genes enhance fertility and survival early in life, but have detrimental effects late in life. These genes are selected in a Darwinian process because their benefits outweigh their costs. Even though they die younger, those individuals carrying the pleiotropic genes leave more offspring, and that’s what counts for evolution. The crux of the theory is that nature is caught between Scylla and Charybdis, forced by limitations of the available genes to choose either high fertility with short lifespan or low fertility with longer lifespan. Crucial to the theory is the assumption that it is biologically impossible to separate the benefits of these pleiotropic genes (fertility) from their costs, so that there is no way evolution could engineer higher fertility without triggering later senescence.
This theory was formulated by George Williams in 1957, long before anyone had heard of epigenetics. He assumed that if you have a gene, you’re stuck with it for life. We can’t blame Williams for the frame of mind that he brought to the evolutionary question, but we now know that this is very much not the case. The body turns genes on and off in individual tissues and at specific times with exquisite precision. In fact, most of the euklaryotic genome is devoted not to genes, but to epigenetic controls of one kind or another.
The fact is that genes are turned on that dial up fertility and promote robust replacement cell growth early in life, and aging at that time occurs quite slowly. Later in life, these growth and fertility genes are dialed way back, and that is the era in which aging comes at us with a vengeance. This, to me, is a direct refutation of Antagonistic Pleiotropy as a theory.
Nevertheless, many examples pleiotropic genes have been found in studies of aging. The above story of telomerase seems to be a conspicuous example. Telomerase promotes epigenetic aging, while lack of telomerase promotes cellular senescence. “If the ’gaitors don’t getcha then the ’skeeters will.”[ref]
My interpretation of pleiotropy is in my book and some of my academic papers. It is this: Aging has been built into our genomes by natural selection for the sake of the community. Fixed lifespan, (especially when modified conditions of food stress) is helpful in preventing population overshoot that can lead to famines, epidemics, and extinction. But whenever a trait is good for the community and bad for the individual, there is a temptation for the individual to cheat (“cheating” is actually the term used by evolutionary theorists). In this case, cheating would mean evolving a longer lifespan via selfish genes that spread rapidly through the population, because they are more successful at the lowest level of Darwin’s competition.
Individual competition would erase aging if left unchecked. The results would be great for individual fitness, but soon would be disastrous for the population. Overpopulation would ensue, followed by the famines and epidemics mentioned above. Evolution has learned (over a very long expanse of time) to protect the communal interest, placing barriers in the way of individual selection for ever longer lifespan. This is the evolutionary significance of pleiotropy. It provides that no simple mutation can substantially extend any aspect of lifespan without adversely affecting another aspect of lifespan or of fertility. The aging clock has been “purposely” configured so as to be spread out over several different mechanisms, tied not just to other pro-aging mechanisms but to fertility as well. Aging is hard to get rid of “by design”.
In the standard theory that I don’t believe, antagonistic pleiotropy is a precondition, and evolution has had to make the best of a bad deal. In my version, antagonistic pleiotropy has been crafted by natural selection in its long-term mode. Limiting lifespan has been so important to the viability of the population that evolution has arranged to protect it from leaking away due to cheating, and antagonistic pleiotropy is one of the ways in which this is arranged. I have modeled this process in numerical simulations of evolution.
My guess is that the connection between telomerase and epigenetic aging is an example of antagonistic pleiotropy in this latter sense–certainly not in the sense of Williams, because on their face telomerase and methylation have little to do with one another.
Bad news for life extension strategies
But whatever the theoretical origins, the pleiotropic connection between telomerase and epigenetic aging complicates any strategy we might devise for slowing the progression of human aging.
I believe that the preponderance of evidence still indicates that activating telomerase has a net benefit for lifespan, but that probably we can add at most a few years by this route. I think that epigenetics is much closer to the core, the origin of aging, and that interventions to modify epigenetic aging will eventually be our holy grail. The caveat is that telomeres are simple, but methylation is complicated, and methylation is just one of many epigenetic mechanisms.
Discover more from Josh Mitteldorf
Subscribe to get the latest posts sent to your email.
Thank You! Is it possible to connect with Bill Andrews for his comments on topic? As i understand right he is one of leaders in field of telomerase activation with tam-818/ta-65 and similar products.
Which one telomerase activator did they use to make epigenetic clock run faster? We know that telomerase is not that body friendly, and giving it in large quantities is not safe, so the preferred pathway is to activate natural mechanism to activate production of it it directly in the cell. But, most of the known telomerase activators so far are highly toxic or even simple deadly for the cell, hence speeding up of methylation clock regardless of any presumed toxicity which exist on telomerase itself (that’s why the cell naturally expresses it on replication only). I’ve recall seeing one study on pubmed on sylibinin (sylimarol), natural telomerase activator from milk thisle, that make epigenetic changes running slower. If one telomerase activator makes running epigenetic changes faster and the other slower, I just would use that that makes Horvath clock running slower. Btw, why Mr. Andrews didn’t joined forces with TA Sciences to replicate their success on putting TA65 through digestive tract to repeat their success on TAM818?
Telomerase is certainly “body friendly” for albatrosses and Blanding’s Turtle (and the stem cells in ALL long-lived animals).
Are you aware of anything pointing toward astragalus being bad for you? Let the Chinese know immediately 😉
OK i see in original text “digested overnight at 4°C with Liberase […] placed face down in a plastic dish with DMEM supplemented with 10% foetal calf serum, penicillin, streptomycin, and gentamycin. […] with 5% carbon dioxide […] Recombinant retroviruses bearing […] the hTERT gene […] mixed with Polybrene […] puromycin was added to kill uninfected cells.” Two separate paths for modified and unmodified cells during creating and later in cultivating.
I had concluded a while back that shortening of telomeres without being rejuvenated by telomerase as we age is an effect of aging and not the cause – I have said so in this blog since long. The paradox referred to in this post can be explained by the onset of inefficiency in our systems to an extent making beneficial things detrimental. My thought on origin of systemic inefficiency is programmed slide in repair vs damage. To be tested in studies whether bringing up repair homeostasis as seen during puberty can resolve the paradox.
I have had the same conclusion with regard to telomere shortening being an effect of aging, existing in some middle realm. I also believe you are referring to Richard Morimoto’s work at Northwestern – which implies that shortly after the onset of sexual maturity, there is a switch that scales down our repair mechanisms.
Yes am a fan of Morimoto.
I saw the telomerase – methylation study and it appears that the presence of telomerase tics off another replication from the Hayflick limit. No telomerase, no tic but shorter telomeres. With telomerase, Hayflick shortens but telomeres don’t. Depressing until you see that Hayflick only shortens during replication. Possible body hack: Don’t take Astragalus every day. Only take it occasionally during times of reduced proliferation such as well into a water fast or under effects of Rapamycin. Opinions?
In terms of telomere length being correlated with Horvath aging doesn’t this ignore the point that Fossel and others have pointed out for some time now that it’s not at all about telomere length, it’s about the rate of shortening of telomeres? Telomere lengths vary widely between individuals so there’s not much of a standard metric for healthy length, but there is (at least according to Fossel) in terms of the rate of telomere shortening. So might it not be a serious red herring that telomere length correlates with Horvath clock aging?
Another problem is using leukocyte telomere length which are only correlated with the health of the immune system and circulatory system. If we could measure telomere lengths of stem cell compartments across organs we might find a different story. Agree with your point about rate of shortening.
Thanks Mark. Could you share your thoughts on telomerase therapy in general? I’m doing a deep dive into this stuff now and am looking for all the good info I can get. I just finished Fossel’s book and I found that his optimism about affordable one hour IV treatments for rejuvenation being available by 2025 wasn’t well supported by the data and studies discussed in his own book, but I’m also finding a lot of other research that in some ways seems to support real optimism — and much that doesn’t. I’ve reached out to Fossel but no response so far.
I don’t agree with others here that telomere length is downstream of other aging effects. Yes telomere shortening can be accelerated by things like ROS and inflammation, but very broadly speaking I think shortening telomeres are upstream of all other causes of aging. You can see this in the evolutionary past as the first aging system to evolve, and i suspect as more studies are done, we’ll find slowed tissue replacement due to shortening telomeres to be behind it all. Every other aging theory needs a more upstream explanantion for why it happens. Telomere theory of aging ties in well with mTOR, epigenetics, Reactive oxygen Species, etc., etc. Whether resetting telomeres periodically back to youthful lengths will be sufficient to defeat aging, I don’t know. We may find issues with stem cells epigenetically drifting even with long telomeres. But it is the best and most technologically feasible option available today.
Thanks Mark. Have you written this up in more detail somewhere? Are you a researcher?
Nope, I’m just an amateur. But reading papers on aging is my hobby, and sometimes hobbyists can see things more specialized researchers can’t.
Very, very interesting PhoenixQ.
Like you I suspect MLTL are a red herring and their increase or decrease can be taken with a pinch of salt. Ditto the poor correlation between MLTL and mortality. We need a better, more tissue wide measure of telomere length. I suspect a sample of Mesechymal Stem Cells would do the job.
I also suspect the correlation between telomerase activity (inferred via SNP variants with longer telomeres) and greater Horvath Methylation age might turn out to not be clinically relevant in an age of telomerase gene therapy. Faster growth with limited regenerative capacity equals faster aging. But if sufficient telomerase were expressed to arrest or reverse telomere loss (it isn’t in any human cell apart from the germ line), then this may reverse aging and render the Horvath measure irrelevant.
Great point Mark, and a conclusion I have already come to.
Over the course of two years, I have adopted many anti-aging strategies and watched my mean leukocyte telomere length improve (via TeloYears testing) – from age equivalent of early 40s to late 20s (I am almost 50). I’m in excellent health, but has it made any change in overt signs of aging such as wrinkles, skin elasticity, gray hair, etc? Nope. My theory is that the Fast Mimicking Diet in particular has a marked effect in leukocyte telomere length due to their destruction and subsequent regeneration, which probably is not representative of avg telomere length across other cells.
At the same time I REDUCED by methylation age (tested via MyDNAge.com) so perhaps reducing MLTL via fasting doesn’t increase telomerase – just selectively eliminates cells with shortest telomeres before more are regenerated from stem cells with longer telomeres.
HOWEVER, there are multiple studies (actually reviewed today in the Medical News Bulletin) that have reported that telomerase activity is increased by fasting and caloric restriction, which has also been shown to increase life span in multiple organisms. You would theorize that the methylation clock is not being driven forward by telomerase during these periods or you would see a shortening of lifespan instead. So there is almost certainly more to this story.
Great in-depth review Josh!
Great points, Phoenix. Would you mind if I contacted you via email to discuss in more detail?
Sure Tam, its my favorite subject. You can reach me via todd -at- gowebb.com.
HI there Josh
Glad to see you adding a new puzzle piece to the logic of “the aging is intentionally made hard to get rid of by evolution idea”.. I had made a similar case with respect to the fact that the hormones that are central to reproduction like LH and FSH are also the hormones that drive the aging process in humans after age 50. Designed so that if you get rid of the aging hormones and live longer, you arent going to be reproducing much.
now I would not be so pessimistic about telomerase causing eppigentic aging on the methylation side….I believe that methyation led aging is reversible…you seem to imply it is permanent..(by your pessimism).I bet if you studied telomerase effects on methylation in calcorically restricted animals you would not get this pro aging effect…as they would have an altered hormonal milieu that would likely prevent this…More studies needed!! See my article>>
the-6-changes-in-lifetime-hormone-levels-that-cause-aging-and-how-to-easily-reverse-them
I agree that methylation changes are reversible in principle, but we don’t know how to do it.
Epigenetic reprograming, surely?
I share your disappointment in regards to telomerase stimulation and extending longevity which is why I stopped TA 65 about 2 years ago. It does seem odd to me though that long mean telomere lengths are mainly associated with very rare cancers, ie., pancreatic, brain, melanoma, and even lung in non-smokers. It’s important to remember that a very slight uptick in absolute risk will be reported as a very substantial relative risk in these rare cancers. They’re still rare even in the presumably large number of people with longer than average telomeres, but why does the risk not extend to the very common colon cancer for instance.
I continue to believe that if you take those with long mean telomere lengths and look at their distribution curve, you’ll find that a very small percentage of them have a heterogenous appearance of too many very long and also very short telomeres putting them at risk for some of the rarer malignancies. But this is still better than having short telomeres and being at risk for the number one killer which is cardiovascular disease.
However, I still think that if one has the lofty goal of say living to 150 and beyond, the problem of telomere attrition will need to be dealt with one way or another. But for now we have rapamycin.
What’s your experience with rapamycin been so far?
All around improved. I’ve lost about 15 lbs., better endurance with much shorter recovery times , feel generally younger, even better mood. No comparison. I just hope that they never stop making it!
Where do you get it from?
I get them online from a place in India called offshorecheapmeds. Costs me about 300 US dollars for 100 of them. Seems reliable but they need an nitial prescription.
Another point to bear in mind is the fact that what is useful from an evolutionary point of view, i.e. minimize any loss due to cancer during fertile years, is not what is best for an aging individual. If an aging individual could theoretically lengthen their telomeres back to what they were at age 20 they might increase their chance of certain rare cancers by a small amount – which would be unacceptable to evolution – but you’d massively reduce your chance of dying overall.
So if an effective telomerase treatment becomes available I know what I’ll do.
Paul if we can maintain homeostasis especially between repair and damage at levels seen at puberty the telomeres will also be maintained via telomerase automatically. Look at how efficiently cancer does this – sufficient to make the cell immortal. This may sound disturbing but I am in awe of the immense engineering power of cancer. What evolution took a million years to accomplish cancer in a tiny fraction of time brilliantly uses available resources of our own body to protect itself from our immune system or poisons like chemo and create immortal cells. What if the secret of immortality lies in the study of cancer? All our research is focused on killing cancer cells. May be we should be doing research in studying the various technologies, mutations and adaptations it uses so rapidly to create immortal cells.
Interesting points.
Thanks Tam
That”s a great point Akshay. Cancer is so intimidating that we tend to ignore how amazing those cells actually are.
Thanks Paul
What is interesting about cancer is that even though it has access to telomerase, it only maintains short telomeres. This I believe is because cancer cells with short telomeres mutate faster – allowing them to adapt to the various challenges to their survival they encounter. Cancer cells truly are the product of survival of the fittest, with most cells dying along the way.
But this also suggests that we should always avoid short telomeres, to minimize the strength of selection on rogue cells.
Well we’ve seen that even genetically identical organisms can differ in lifespan by 10 fold with naturally occurring tweaks to gene expression(some social insects). So the idea that there is programmed aging, and probably interlocked mechanisms to make any lone required mutation harmful is sound.
As we’ve seen changes within and between species, it seems most of what is done by nature is changes to gene expression not novel exotic proteins to solve engineering problems.
That said, we also know there are negligible senescence organisms. The longer lived a species, I believe, the closer it must be to reaching a state of negligible senescence. While it may hypothetically take multiple simultaneous mutations to change gene expression in such a way to enact such a transition, for a longevous enough species one might imagine a hypothetical drug cocktail could affect the various pathways in the right way just enough to allow for such a transition to take place.
A very interesting post.
1. Wonder if a staged therapy that first lengthens telomeres, and then helps to remove the older methylation patters is possible.
2. You mentioned that you’ve modeled (antagonistic pleiotropy as a more deliberately evolved trait) process in numerical simulations of evolution. – Can you link us? I’d love to see your code/paper/etc.
Josh and I have always been of a mind about aging, it’s evolutionary import, and that it is a species characteristic, like size or mating times that are an integral part of a species fit to its niche. This latest report tends to bolster that impression, an organism tends to protect it’s lifespan as it does other species characteristics, in this case by a clever stratagem Josh characterizes as a Catch-22 or “if the ‘skeeters don’t get you the ‘gators will”. That seems to put a damper on the life-extension exercise, but I think that the current thinking about life extension is incorrect in that real life-extension by the current thinking, of limiting damage or repairing it by exogenous agents is wrong-headed and would lead to an exacerbation of current problems, maybe putting them off for a while, but ultimately leading to even larger and more long-lasting ones. Simply, those current attempts (not mine or my group in Mumbai) will lead to a lengthening of the periods of senescence – as life histories seem to all follow a similar trajectory, in that a lengthening of life means a lengthening of senescence a period marked by decreasing inability and decreasing ability.
The royal road to life extension is through rejuvenation; there is not enough time or space to state all my reasoning, but suffice it to give but one example of this phenomenon at the cellular level (though I don’t think aging occurs at the cellular level, but ALSO occurs at the cellular level. The example is one that contravenes the newly discovered principle of telomerase causing Horvath aging while stopping Hayflick aging. It is simply (and this was demonstrated by Laure Lapasset) that if cells are induced by Yamanaka factors to pluripotence, not only do their telomeres increase in length but the all aspects of their structure and behavior reverts to a young age phenotype. So the answer to life extension, the answer we are actively pursuing is not to extend life by reducing or even repairing age-related ‘damage’ (at least not repairing such damage by exogenous means as the SENS foundation would have us believe) but to direct the cell to repair its own damage by rejuvenating it – having return to a young phenotype. Now of course there’s the problem, we wouldn’t want our cells to revert to embryonic phenotypes – we’d become a plastic messes, giant teratomas, and surely wouldn’t appreciate that. Tom Rando of Stanford realize this and asked, in one of his publications whether the induction to a younger age-phenotype couldn’t be accomplished without cellular de-differentiation? I answered him (in an e-mail) that he and his group (consisting of the Conboys, and Amy Wagers among others) had already shown that it could in their 2005 ‘letter’ published in Nature (Rejuvenation of aged progenitor cells by exposure to a young systemic environment.) He already knew about my heterochronic plasmapheresis idea and he soon agreed that he’d put someone on the case (just before Wyss-Coray, his subaltern started injecting ‘young blood’ plasma into mice – coincidence – I don’t think so).
So my point is that, if done correctly, (which I think it was/is not), the body has the potential to change the epigenetic marks characterizing and possibly causing aging while at the same time lengthening telomeres to youthful levels. In fact, Lapasset’s work showed that all of the putative causes of aging, ROS production, mitochondrial inefficiency, even the transcription profiles (almost certainly the result of epigenetic changes) could be returned to a youthful phenotype. My analogy is to curing measles, we can apply ointments to stop the rashes, aspirin to lower the fever and cold baths to bring down the temperature – but we’re only ‘curing’ the symptoms and not the cause. We are now trying to ‘cure’ the cause of aging (a biological program, a continuation of post-adult development that Nature, clever as she is, has left us a loophole in. We are pursuing that strategy with a twist that will make this process into one affordable to everyone who needs it, and eventually, that’s all of us.
Your theory makes a lot of sense to me. However at this day and age, taking plasma from a young person giving it to the old is considered extravagant if not immoral. I guess that’s why you are pursuing it with a “twist”. That “twist” will not only make it cheaper, but acceptable in regular people’s mind. Am I right assuming that?
Well, I wish you early success.
Man, I wish that that I could go back and proof-read what I just wrote, so many errors (but not in thinking) – but once you’ve hit submit – story told!
I believe the answer is that a cell knows how long its telomeres are and uses this as a metric to decide what rate of growth to set. So telomerase is a sort of growth hormone. More telomerase, more growth. Therefore as telomeres shorten the cell slows down both its internal turnover of proteins, and its replication. This means we can survive for longer with a limited supply of telomerase.
OK, I worked in the Shay-Wright telomerase lab for five years, so I am a wee bit biased. But I immortalized cells using telomerase back in 2001 that are still in culture. From over a hundred species.
Somehow the magic methylation clock doesn’t stop them… their brother cells stopped dividing after 80 divisions, and yet the telomerized versions never stop.
I’ll read the paper, but giving up on telomerase activation makes little sense to me.
Speaking of epigenetics, the nicotinamide riboside Phase 2 study is publishing any day now.
‘Somehow the magic methylation clock doesn’t stop them…’
EXACTLY!
Hi Bill, do you think that the telomerase immortalized cells are functionally young after so many passage?
Being able to replicate indefinitelly may be one thing, being functionally useful in the tissue context is another.
I have read several studies from the early 2000s on telomerase immortalized cell lines and it seems like they are high on growth hormones which does not happen naturally, plus they evolve to overcome the p16 mediated senescence by epigenetic means.
So that in some cell cultures there is a crisis happening at about the Hayflick limit after which only p16 hypermethylated cells duplicate.
Didn’t Horvath show that the cells ‘aged’ epigenetically in vitro just as they would in vivo, only much faster as you would expect due to culture conditions? To me this suggests the process is reliable and repeatable and those cells were normal aside from the growth stimulating effect of telomerase. As I’ve stated above I believe cells know how long their telomeres are and this is how they regulate their internal rate of molecular turnover, as well as their speed of division when this is permitted by serum conditions. Therefore telomerase immortalised cells are just set to grow at maximum rate. I expect this is what drives the acceleration of the Horvath clock.
This is probably irrelevant to potential life extension treatments however, as proposed treatments use a plasmid to temporarily express telomerase to elongate telomere back to youthful levels, rather than have it permanently turned on. This should reset cells back to youthful metabolism. The onus is on Horvath to show how this could cause harm. So far his clock is just a clock.
Cells in culture are selected for faster growth. And the conditions are very unnatural, O2 is far too high, hormone levels all wrong (they are grown in cow serum regardless of their species… nobody wanted to get the mouse OR whale serum).
Shay-Wright does do some low-O2 culture, so we had some normal vs. telomerized cells in a slightly more normal environment.
But the chromosome numbers are stable. Telomerized cells are no more unusual than other normal cells in culture. And they never slow down dividing… there is no methylation clock in telomerized cells in culture.
You should get Horvath to run his clock on some of the cultures you immortalised back in 2000 and give you an age. That would be good for a laugh.
Hey, it was only 2001, I’m not that old 😉
Yes, that would be a useful experiment. But I think the changes would occur quickly as the cells went into culture and then level off to whatever methylation pattern gives faster growth… cell culture selects rapidly for fast growth, and when you’re talking 17 years of cell culture that’s a lot of generations at two doublings per week.
Well according to this 2016 paper by Horvath: ‘Epigenetic clock analyses of cellular senescence and ageing’, immortalized cells continued to ‘age’ epigenetically from passage 7 to passage 50, when the experiment was concluded. I assumed this would continue ad infinitum, but I haven’t seen data on that.
I suspect Horvath would find the same thing Michael Rose’s lab at Irvine found -that at some point aging stops. There is likely some end point to the methylation changes so the Horvath clock would cease being an accurate predictor for cells of extreme chronological age.
That’s a very interesting point. Do you have any further information on Rose’s work I can read up on?
Also given Horvath has calibrated his measure to chronological age, it would probably lose all meaning at ages of 100+, not least because he wouldn’t have the statistical power for an accurate age determination.
Great info. Can you share any data/preprints/published studies on this?
Hi Bill,
thanks so much for sharing your first hand experiences. I have contacted the researcher who published the paper on the accelerated Horvath clock in telomerase immortalized cell line and he said the high passage cells looked different morphologically he just didnt have time to find out why.
Thats why I asked about the youthfulness of the immortalized cell lines.
Again this is interesting, but not really relevant to a potential treatment to re-elongate telomeres back to a youthful length. Here we are not immortalizing, just reverting gene expression levels and the metabolism of the targeted cells. Theoretically the temporarily elevated telomerase levels might accelerate the Horvath measure of aging. But this measure is calibrated to normal aging and probably isn’t relevant in this case.
I think the technology is there to find it but I doubt it could be approved by authorities let alone finding brave volunteers.
You take stem cells from your own bone marrow – there are established procedures for this.
You then genetically engineer them to grow telomeres. Partially kill your bone marrow with low dose chemotherapy then take the genetically engineered stem cell transplant.
I think all parts here are well established protocols. These are used in CAR-T and in immune system reconsttruction after chemotherapy.
If your theory is correct you end up with a younger better immune system,
For a younger immune system one could also take LDN and reverse the Involution of the thymus. Probably same result with less pain.
LDN has been shown to reverse Thymus loss?!?
No Mark as you may know they improve immunity by different paths. LDN improves immune function by its hormetic upregulation of OGF-OGFr system – endorphins enkephalins. OGF has shown anti inflammatory action. Separately many studies from 3 to 4 decades have shown that regrowth of Thymus is possible and improves immune function. Dr. Greg Fahy had conducted a FDA IND study on 9 healthy human males and demonstrated regrowth of Thymus is possible. It seems parabiosis is unable to reverse Thymus involution. If one goes by past mice studies then a synergistic cocktail of easy to purchase vitamins and minerals may also be able to regrow the Thymus.
NOTE added by JJM: Greg’s trial with growth hormone may not have been successful – it’s been several years, and he hasn’t reported results.
That’s an interesting idea GaborB. Immune System has been totally replaced in a few patients with good results. But doing the same to all stem cell compartments in the body could be even more tricky.
Josh, did you study how widespread mitotic telomere shortening is among unicellular eucaryotes?
Are there only a few species with this strategy or more?
It happens in slime molds. (My undergrad thesis from 2001 😉
I don’t know how widespread it is. I don’t know if anyone knows.
Stem cells and gametes have high telomerase expression yet a low DNAm age. How? Telomerase by itself is probably not sufficient to increase the DNAm age, not in every cell type. Other mechanisms must be involved.
The cell culture they did only involved fibroblasts from neonatal foreskin. Given that embryonic and neonatal tissues already have a rapidly advancing DNAm clock, I can’t help but wonder how relevant this is to adult cells.
Could you flesh out your thoughts on this a little more?
Telomerase is associated with regeneration, not with lifespan. I agree that tissue involution could increase mortality risk, but there are way too many short-lived species where telomeres don’t shorten with age as they display indeterminate growth, meaning they don’t stop growing in size once they reach adulthood.
There is the Myotis bat that has 21 telomere linked maintenance genes, 14 for DNA repair and 5 for alternative telomere lengthening mechanisms, and they live an incredible 35 years. Their telomeres do not shorten as they age and they are no bigger than a mouse. That certainly seems to support longevity via an ALT mechanism of telomere size maintenance,
Bats live longer than mice because they are able to fly so they can escape predators much faster. The naked mole rat is a rodent which lives around 30 years as well – do you know if its telomeres are shortened with age or not?
Or rather the fact they can fly and escape predation has allowed evolution to select for longer lifespan. The story is the same with NMRs but instead of flying the adaptation that allows this selection is living underground.
‘Genome Stability Maintenance in Naked Mole-Rat’ shows that NMRs have telomeres of a similar length to humans rather than normal rats, but that genes involved with telomere maintenance have been positively selected for. It doesn’t say whether they shorten over their life or not, but given that it is now reckoned NMR mortality does not rise with age (see the paper from Calico ‘Naked mole-rat mortality rates defy Gompertzian laws by not increasing with age’), I have to assume that they do not. Its a worthwhile experiment for somebody to do.
I’m sure there is a Shay-Wright paper on this, we had NMR cells.
Hi Bill,
This 2007 Aging Cell paper ‘Telomerase activity coevolves with body mass, not lifespan’, suggests NMRs have high telomerase activity (measured across 6 different tissue types) and do not use replicative senescence as an anti-cancer mechanism, yet they suffer very little cancer. It would be interesting to see what their epigenetic methylation clock looks like.
Naked mole rats are pretty amazing actually and recently it’s been found that they don’t actually age, at least not like any other mammal, and certainly not like we do. In humans the risk of dying doubles every 8 years after the age of 40 but in the NMR there’s no increase in the risk of death even when they are 25 years beyond puberty. They are virtually immune to all age related illnesses and their death is completely random regardless of their age. On any given day their chance of dying is about 1 in 10,000. Also strange is that in captivity the cause of death is almost impossible to determine even with an autopsy. Usually all that is found are mouth ulcers.
To be honest I’m more interested in their mTOR levels than I am in their telomeres and I’m wondering if they have some natural way of downregulating those levels as they grow.
Paul – The part about “constant mortality” independent of age is new to me. If this were literally true, we would expect one individual in 1,000 to live for 200 years. To my knowledge, the longest-living NMR on record is 35.
http://www.sciencemag.org/news/2018/01/naked-mole-rats-defy-biological-law-aging
A description I’ve heard more commonly in the past is that they don’t age and don’t age and don’t age and then they suddenly drop dead. Some sea birds have the same mortality pattern.
I find it hard to believe that NMR would just drop dead. Death would need a cause – a failure. Our biological systems are so robust in their prime that so far not come across any studyi showing evidence of death without some system failure. Disease, mutation, accident or predation is more likely.
The Calico paper was pretty thorough in showing mortality of NMRs didn’t rise with age. But I don’t think it was as low as 1 in 10,000. Because they live so long it’s unsurprising we don’t have reliable numbers on this.
Hi Anca,
Specifically, can you name any short lived species of animals that do not stop growing and yet do senesce?
Thanks Anca,
Kevin Brown
Sure.
Here are 3 species of sea urchins with different lifespans, even if they all maintain their telomere length and there is no decline in regeneration with age in either of them:
3-4 years – the Lytechinus variegatus sea urchin
> 50 years – the Strongylocentrotus purpuratus purple sea urchin
> 100 years- the Strongylocentrotus franciscanus red sea urchin
References:
Francis, N., T. Gregg, R. Owen, T. Ebert, and A. Bodnar. “Lack of Age-associated Telomere Shortening in Long- and Short-lived Species of Sea Urchins.” FEBS Letters 580, no. 19 (August 2006): 4713-7. doi:10.1016/j.febslet.2006.07.049.
Many primitive aquatic multicellular animals grow continuously and many of these species (but not all!) do not age – these animals include corals, sponges, sea urchins, some bivalves.
One colony of corals that dies in a couple of years is Stylophora pistillata, while many coral colonies survive for thousands of years: Colpophyllia natans brain coral 2,000 years and the Leiopathes coral 4,270 years.
References:
Rinkevich, B., and Y. Loya. “Senescence and dying signals in a reef building coral.” Experientia 42, no. 3 (1986): 320-322. doi:10.1007/BF01942521.
Sussman, Rachel, Hans Ulrich Obrist, Carl Zimmer, Christina Louise Costello, and Michael Paukner. The Oldest Living Things in the World. Chicago; London: University of Chicago Press, 2014.
Hydras stop growing once they reach maturity so they are determinate growers, but even here there are species which do not age (Hydra vulgaris) and species which age (Hydra oligactis).
References:
Martı́nez, Daniel E. “Mortality patterns suggest lack of senescence in hydra.” Experimental gerontology 33.3 (1998): 217-225.
Yoshida, Kengo, et al. “Degeneration after sexual differentiation in hydra and its relevance to the evolution of aging.” Gene 385 (2006): 64-70.
Hi Anca,
Thank you for your reply, this has been an area of my interest for about 10 years now.
It seems that many of the animals you named that both have a finite lifespan while showing indeterminate growth also reproduce asexually. Do you know of any animals that are obligate sexuall reproducers that show indeterminate growth and still senesce? From an evolutionary pressure perspective the lifespan of an asexual species should be measured from the start of the clone until that last member of the clone dies. If you think about it asexually reproducing animals can be seen as one organism with redundant organ that has given up contiguous contact between redundant parts to increase the probability of survival of the clone over time. Just as we use apoptosis to destroy cells for the greater good of the large organism The clone can sacrifice individual members of the clone without sacrificing reproductive capability of the clone sexually or asexually.
Are any of the example organisms you provided, unable to reproduce asexually?
Thanks Anca for sharing your knowledge about this. I have proposed the theory that aging is a sexually selected trait, therefor species that do not select their mates such as one that release eggs and sperm into the water should evolve toward longer life span (non-senescing) over generational time. So I was surprised when you described species that aged while being indeterminate growers.
Hi Kevin,
I’m also very interested in comparative gerontology ever since I first wondered whether all species age and if not, whether this could be a source of inspiration for novel anti-aging therapies just like flight in birds was first the inspiration for airplanes (which got better at it in the meantime).
To answer your question, Caleb Finch mentioned some rockfish and cod species as displaying negligible senescence (source: Finch, Caleb. Longevity, Senescence, and the Genome. Chicago: University of Chicago Press, 1990.)- fish reproduce sexually, but I’m not sure whether these specifically grow indeterminately or not.
But you may want to check on the Bidder’s hypothesis which was refuted since 1921 – he stated that aging starts when growth stops.
As mentioned above, Hydra vulgaris doesn’t grow indeterminately, it is able of undergoing both asexual and sexual reproduction and yet it doesn’t age.
The Guppy fish (which you can find in most aquarium pet shops) grows indeterminately as female and determinately as male, but ages gradually.
References:
Arendt, J. D., and D. N. Reznick. “Evolution of Juvenile Growth Rates in Female Guppies ( Poecilia Reticulata ): Predator Regime or Resource Level?” Proc Biol Sci. 272, no. 1560 (February 2005): 333-337. doi:10.1098/rspb.2004.2899
Reznick, D., M. Bryant, and D. Holmes. “The evolution of senescence and post-reproductive lifespan in guppies (Poecilia reticulata).” PLOS Biol 4, no. 1 (January 2006): e7. doi:10.1371/journal.pbio.0040007
Well Anca, humans have both continuously growing tissue that suffers from limited regenerative potential, i.e. vascular aging, immune aging, skin aging, and quiescent tissues that also age through accumulation of faulty mitochondria, impaired lisosomes, etc. It is not yet clear which, if either, is primary, but my bet is on the proliferative tissue.
Anca
I think that it may be true that telomerase is not associated with longevity in a direct cause and effect manner, but certain agents like rapamycin,ashwaghanda, white willow bark, etc., will only get you so far and then the issue of DNA instability secondary to critically short telomeres will rear its ugly head.
I do acknowledge a certain risk in an induced , blind , telomerase stimulation self experiment , especially the possibility of very rare cancers as mentioned in Josh’s post, but there is every reason to believe that rapamycin would negate such a risk.
Senator Johnson’s “right to try” bill just passed. Many states (including mine, where I helped pass the bill) have such laws… now maybe with the Feds out of the way, it will be a lot easier for terminally ill people to try something that might work.
Good job!
” identified a set of 353 methylation sites”
Which I was sure you’d be breaking down into their appropriate categories, over the next few posts..
What held you last month? the ‘gaitors or the ‘skeeters? : )
NMR’s are certainly not immortal but they are the only known mammals to not have an increase in the probability of death as they age. So the chance of them dying is the same at 25 years old as it is at 1 year old.It would be as if humans were equally likely to die at 20 as they are at 90.
Of course they do die, but the pattern is more like exponential decay where the half-life is 19 years. They are virtually immune to all age related type illnesses like CVD, cancer, Alzheimer’s, etc., and like I said those in captivity usually just show numerous mouth ulcers as if their buccal defenses had broken down and they got some sort of infection.
So I’ve been searching the literature today in an attempt to support my new found belief that the naked mole rat has a pattern reminiscent of the One induced by rapamycin. Indeed this has been studied https://www.biorxiv.org/content/early/2017/09/21/191999. The abstract states : The set of 319 positively selected genes contain regulators of mTOR and is enriched in functional terms associated with 1. Processes that are regulated by the mTOR pathway, eg, translation, autophagy, and mitochondrial biogenesis, 2. The immune system, 3. And antioxidant defense. Analyzing gene expression of PSG’s during aging in the long lived naked mole rat and up regulation in the short lived rat, we found a pattern fitting the antagonistic pleiotropy theory of aging.
“ Long lived rodents reveal signatures of positive selection in genes associated with lifespan and eusociality”. Arne Sahm. 9/21/17
So these little monsters have their own built in rapamycin system which immunizes them against all age related illnesses and gives them an extraordinary lifespan, but it’s the mouth ulcers and secondary infection due to the buccal proliferation problem that ultimately kills them. It all fits.
Interesting. But NMRs also have high levels of telomerse, unlike humans.
Yes, they use the ALT pathway, so they get both TOR inhibition as well as telomere maintenance and live a very long and disease free life. Probably both are necessary.
May the TOR inhibition prevents the small propensity for the rare cancers.
Does anyone know how the ALT path to telomere extension actually works?
https://www.ncbi.nlm.nih.gov/books/NBK6486/?report=reader
Pretty much sums it up.
Interesting that telomeres in the ALT mechanism use the telomere from another chromosome as a template. Still don’t understand how this actually allows such long telomeres though. Frustrating that this has not been better elucidated.
The more I think about it, the more convinced I am that humans will need to restore long telomeres to live to great age in good health. It may not be a consistent pattern across species, but as we’ve seen time and again evolution has used many different mechanisms to achieve longevity. But for humans a lack of regenerative ability is clearly a bottleneck.
This was an interesting discussion, thanks.
Interesting.
How much overlap those 319 sites would have with the 353 from Horvath’s clock?
Almost certainly none. The Horvath changes have no effect on gene expression as far as I know.
There seems exist an alternative pathway to lengthen Telomeres without enhancing telomerase activity.
This paper from January 2017 (University of Vienna) is mentioning it:
https://www.researchgate.net/publication/313063871_EGCG_Containing_Combined_Dietary_Supplement_Affects_Telomeres_and_Epigenetic_Regulation
There are two ways to lengthen telomeres, telomerase and ALT. Other methods can slow the rate of loss, but can’t lengthen them.
https://www.researchgate.net/publication/8556361_Bechter_OE_Zou_Y_Walker_W_Wright_WE_Shay_JW_Telomeric_recombination_in_mismatch_repair_deficient_human_colon_cancer_cells_and_telomerase_inhibition_Cancer_Res_64_3444-3451
Hi Josh! I am a respectful follower of your articles and blogs, and would appreciate your views on the present media hype regarding the efficacy of nicotinamide mononucleotide in reversing muscle loss etc in old age. The mouse studies are interesting, but what is your view on extrapolation to ourselves? Thanks!
Ditto, and while you’re at it Josh, what are your thoughts on NMN vs. NR? Phase 2 study on NR is coming out in Nature soon, that should add some data there.
Oh, and are you follwing the preclinicals on imetelstat + venetoclax in AML? Or the imet trials against MF and MDS? I’m always talking about telomerase activation, but the anti-hTERC effect of the imetelstat oligo looks like the next big thing in anti-cancer combination treatments.
I know nothing about these trials. So many things to follow up on, I can’t promise to cover this.
In support of that, Geron’s stock has been multiplied by 3 in the recent weeks. It looks like many people are expecting the data of the iMbark trial for MF (which should completed in the next few months) to be really good.
http://www.geron.com/patients/clinical-trials/
However, Geron has a long history of hope and delusion so I am waiting for the data before acknowledging the potential of imetelstat. Hopefully, it will bring good news for patients with MF.
Geron gave up on telomerase activation and gave it to Andrews’ Sierra Sciences for a song. So I’m no fan of their CEO’s strategic direction… but imetelstat is working in MDS too (see the recent presentation by Dr. Raza), not just MF. And it worked in ET, in an old trial (an NDA was not pursued as “not commercially worthwhile”). And the preclinicals with venetoclax in AML presented at the last ASH look very promising… The clinical mechanism of imetelstat is not telomere shortening, that confused the issue for years.
Very interesting. Looking forward the next trial results.
Actually I saw that Shay and Wright have published the paper “Telomerase Inhibitor” in 2001 so maybe you know why imetelstat is the only one that is currently used in cancer (up to my knowledge). What is special about it?
Imet doesn’t just inhibit telomere lengthening. It inhibits the hTERC RNA and all its signals.
BTW, the Phase 2 trial on nicotinamide riboside just published in Nature Communications today.
Interesting.
Thanks
Does anyone know if anyone has applied the Horvath epigenetic clock to compare M. Rose’s long lived fruit flies to the M. Rose’s Control group flies? This could possibly help answer the question about what accounts for the plasticity in the rate of aging that we see in such short evolutionary time. It seems that this could shed light on whether mutation or epigenetics more strongly accounts for the plasticity we see in the rates of aging for example in possum populations isolated away from predators on islands such as work done by Steven Austed.
Hi All,
Does anyone know if anyone has used the Horvath epigenetic clock methods to compare M. Rose’s long lived flies to his control flies? It seems to me that this could shed light on the question of whether aging is more a transgenerational epigenetic phenomena or whether a slower rate of aging in these flies is the result of mutational events?
Like Josh, I will bet that the evolutionary speed of adaptive plasticity in the rate of aging that we see in nature is primarily the result of the transgenerational execution of epigenetic programming extending or shortening life spans in offspring in order to adapt to environmental change, and is not primarily the result of mutations.
I think, as a community, we often underestimate the complexity and capability of the regulatory programming both genetic and epigenetic that governs eukaryotic life. Without the experience of developing complex software systems, most people greatly underestimate how sophisticated and nuanced regulatory control can be in complex systems be they software or biological. As a result we too often default to the idea that mutations are required to elicit the behaviours that control the lifespan of species in nature.
Hi Josh,
in your article “Telomerase- Update and Downgrade” you state “I have modeled this process in numerical simulations of evolution”. Can you tell me if that is published and if not did you use Genetic Algorithms in your approach? I have done a lot of work in GAs and have always felt that it would be quite useful in elucidating the cell and its epigenetics etc since the method thrives on many variables in order to approach optimum solutions.
One other thing sort of related, some time ago I asked a question attempting to cast light on some differences between the work of dePinho (Jaskelliof) and Blasco in extending the life of mice. After some thought I came to a tentative conclusion that Blasco’s application of telomerase was of the single injection variety, resulting in very small life extension (10%-25%), whereas dePinho (Jaskelliof’s) work was of the continuous production of telomerase by the extra gene inserted into the DNA. This was probably significant in that apparently the cell will only effectively apply telomerase to the telomere during S-phase and not continually. This thereby greatly lessens the chances of telomere extension (to say a few hours every year in the case of humans). In Jaskelliof’s work the mice were (apparently) completely rejuvenated but reportedly did not survive longer than normal (killed off because of the problem with the terms of the grants from the US source? in not supporting work in aging?).
Frank
Hi Josh
I had another thought about this relationship between telomerase and epigenetic age. Actually I got the idea from something Ed Park said.
What if epigenetic age is just a measure of time since a somatic cell line was spawned from it’s stem cell progenitor (which by definition will be younger epigenetically)? That means older people would naturally have an older epigenetic age because their stem cells are less active and somatic cells are replaced less often. It also means an individual with sightly more telomerase (and longer telomeres) would naturally have longer lasting somatic cells, so at any given time their tissues would be older epigenetically because they are also replaced less often. If this is true it means there are two separate but easily confused correlations: older people having less often replaced somatic cells due to tired stem cells, and people who have longer telomeres needing replacement less often (but probably with better preserved stem cells as a result).
Please let me know what you think.
Oh and I should have said – it is the people with longer telomeres at any age that give rise to the correlation of more telomerase: older epigenetic age. But this doesn’t mean these people are older biologically, it only appears that way because like people who really are older biologically, they replace somatic cells less often.
I really think this is the answer.
It is also related to my explanatiin for why these people have slightly more rare cancers: it is easier for an emerging cancer to produce more telomerase in someone who naturally has more of it (but note this will still require some short telomeres in my opinion).
Finally, the telomerase-epigenetic age signal should be smaller in tissues that renew more slowly, although I would expect it not to disappear, as telomerase can protect cells in times of stress. Something to ask Horvath to look at in his data, perhaps?
Interesting study here on Nicotinamide riboside that claims supplementation with mimics calorie restriction.
https://www.nature.com/articles/s41467-018-03421-7
When I read the above mentioned paper I only see two conclusions the authors have written: 1. 1000 mg of NR a day is well tolerated. 2. NR is promising in lowering SBP an aortic stiffness.
I went back to read Josh’s 2014 post “Nicotinamide Riboside – where’s the beef?” and noticed in the discussion that there were a lot of people who raved about how well they felt after talking NR. Now 4 years later, I wonder if they continued taking NR and how do they feel now. Would love to hear their comments.
Here the claim for CR mimic is made more explicitly in quotes from one of the authors.
http://www.sci-news.com/medicine/nicotinamide-riboside-05864.html
I would like to know if Josh has a different take on NR now and with you would like to know other people’s experiences.
I hope I am able to change my mind when I get something wong. I’m changing my view about telomerase. But in the case of NR, I see no reason to change my position. There is a lot of hype and money promoting NR, but only secondary indications of its benefit. I’m waiting to see evidence that it lowers mortality rates in humans or extends lifespan in rodents.
I see a new criterion on the horizon–in fact I’m advocating for it. That is retarding the methylation clock. None of the life extension treatments have been tested yet by this standard, but it’s relatively cheap and convenient compared to human clinical trials or even animal studies. I’d like to know whether NR can slow the Horvath clock.
But what do you think of my idea that the reason telomerase and epigenetic age are inversely correlated is because longer telomeres mean somatic cells need replacement less often? This would resolve the paradox rather well, don’t you think, and is testable?
It also means that if you could increase cell turnover you’d decrease epigenetic age, because new somatic cell lines would be closer epigenetically to their progenitors, but this might also exhaust stem cells. So not everything that reduces epigenetic age is good necessarily.
Thank you Josh for clarifying your position.
Some people at the longecity discussion forum take 4 grams a day, that’s over $600 a month for a single supplement! If I read between the lines, it looks like it might benefit overweight people, hence CR mimics.
The Epigenetic Paradox of Pluripotent ES Cells
Very interesting review paper. they claim that epigenetic age 0 cells – ES cells from blastocysts – exists in an epigenetic control free state. These cells can maintain their 0 age for an infinite number of divisions and they elongate their telomeres via mechanisms related to meiosis. As if they were from some precambrian protozoa. As soon as gastrulation begins they start to rely heavily on epigentice regulation like histone modifications and DNA methylation. So all this epigenetic development and aging is an added layer on top of the unicellular eucaryote.
I have recently become more skeptical about NR, even though I purchased some myself. The science media is all a buzz right now about boosting NAD+ with Nicotinamide Riboside. Several studies have just come out suggesting this is a health promoting supplement. Boosting NAD+ is supposed to raise Sirtuin levels which have been shown to be health promoting. However, there have so far as I know, been no lifespan increases shown to be associated with boosting NAD+. I am concerned that boosting NAD+ will undermine the proven benefits of energy restriction from CR, Fasting, or pharmaceuticals such as Metformin, Resveratrol, and Rapamycin. Here’s why: All of the proven life extension methods involve reducing energy, slowing metabolism, encouraging autophagy and slowing Mtor. Is boosting NAD+ the same short term robustness vs long term longevity trade-off we see with eating vs fasting. The first study below was just released and shows that boosting NAD+ increases cellular ATP levels. The second and third references show that higher ATP levels will REDUCE AMPK and INCREASE MTOR. Not the way to increase lifespan (so far)!
First study: “Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD+ in healthy middle-aged and older adults
Christopher R. Martens, Blair A. Denman, […]Douglas R. Seals
Nature Communicationsvolume 9, Article number: 1286 (2018)
doi:10.1038/s41467-018-03421-7
29 March 2018
“Nicotinamide adenine dinucleotide (NAD+) has emerged as a critical co-substrate for enzymes involved in the beneficial effects of regular calorie restriction on healthspan. As such, the use of NAD+ precursors to augment NAD+ bioavailability has been proposed as a strategy for improving cardiovascular and other physiological functions with aging in humans”
“In addition to boosting NAD+-specific metabolites in PBMCs, we also observed increases in the mean concentration of other metabolites involved in the regulation of energy production and metabolism, including adenosine and adenosine triphosphate (ATP; mean change = 699 pmol per mg protein; one-sided 95% CI (84, ∞); Fig. 2 and Table 3); however, analysis of this metabolite was considered a secondary outcome and the increase did not attain statistical significance after correction for multiple comparisons. NR supplementation also tended to raise levels of adenosine diphosphate (ADP) and adenosine monophosphate (AMP), though increases in these metabolites did not reach statistical significance (Supplementary Table 4). Collectively, these findings indicate that chronic NR supplementation effectively stimulates NAD+ metabolism in healthy middle-aged and older men and women.
Metabolite Median Median Range Range
Placebo NR Placebo NR P-value
NAAD 0.0 0.0 0.0−2.3 0.0−8.7 0.018*
NAD+ 7.7 12.2 0.0−27.4 4.7−67.8 0.048*
NADP 6.1 6.3 3.3−17.9 2.7−42.7 0.267
NaM 257.6 278.6 109−411 171−1357 0.065
ATP 1592 2205 363−3446 763−5459 0.032
All values expressed as pmol per mg protein. * represents unadjusted P < 0.05; ATP represents secondary outcome assessed at Bonferroni-adjusted P < 0.006"
I'm not sure why the statistical significance level of .05 was not used for ATP but just looking at the numbers it seem clear to me that NR raised ATP.
Second study referenced: "Cell Rep. Author manuscript; available in PMC 2017 Nov 16.
Published in final edited form as:
Cell Rep. 2017 Oct 31; 21(5): 1331–1346.
doi: 10.1016/j.celrep.2017.10.029
PMCID: PMC5689476
NIHMSID: NIHMS913322
PMID: 29091770
The mTORC1 signaling network senses changes in cellular purine nucleotide levels
Gerta Hoxhaj,1"
“…mTORC1 signaling is also sensitive to changes in cellular energy status. A primary mechanism by which mTORC1 is suppressed by energy stress is through the action of the AMP-activated kinase (AMPK). AMPK is activated by a rise in the ratio of cellular AMP to ATP that accompanies excess ATP consumption or decreased ATP production through glycolysis or mitochondrial respiration (Hardie, 2014). AMPK suppresses mTORC1 signaling through both the TSC complex and through direct phosphorylation of the mTORC1 component Raptor (Gwinn et al., 2008; Inoki et al., 2006; Inoki et al., 2003)…”
Third study referenced: Cell Metab. Author manuscript; available in PMC 2017 Nov 17.
Published in final edited form as:
Cell Metab. 2014 Dec 2; 20(6): 939–952.
Published online 2014 Oct 30. doi: 10.1016/j.cmet.2014.09.013
PMCID: PMC5693325
EMSID: EMS74925
PMID: 25448702
"AMPK – sensing energy while talking to other signaling pathways
D. Grahame Hardie
…The AMP-activated protein kinase (AMPK) is a ubiquitous sensor of cellular energy and nutrient status in eukaryotic cells. Because of the adenylate kinase reaction (2ADP ↔ ATP +AMP), which appears to be maintained close to equilibrium in essentially all cells, increases in the cellular ADP:ATP ratio (signalling a deficit in cellular energy), are always accompanied by even larger increases in the AMP:ATP ratio (Gowans et al., 2013). AMPK monitors the ratio of AMP:ATP (and, to a lesser extent, ADP:ATP) and, if energy deficit is detected, acts to restore energy homeostasis by switching on alternate catabolic pathways that produce ATP, while switching off biosynthetic pathways and other non-essential processes consuming ATP…”
So my question is – does taking NR negate the positive effects of CR, Fasting, Protein Restriction, Exercise, Metformin, Berberine, and Rapamycin?
You make a great point Neil. No evidence of longevity with NR. The users state that it gives them more energy, but in and of itself that’s not much of an anti-aging endorsement. I can get more energy from steroids, growth hormone, and even amphetamines, but none of them are longevity promoters, and as you say, the anti-aging agents tend to slow everything down.
I wouldn’t have thought NAD boosters negate CR or mTOR inhibition – in fact I suspect higher ATP is downstream from better mitochondrial health, so this is probably exploiting the same or similar pathways.
This is yet to be proven of course but I think we’ll find NR or NMN has far greater an effect on health span than lifespan in humans, i.e. it will increase the average but not change the max much.
Hi Neil,
To my knowledge nothing, CR, metformin, rapamycin, etc has been shown to extend human maximum lifespan? So to me, it is speculative that anything like NR or NMN can be distinguished from these other interventions at this time. They all can be seen as modulating health span.
If Josh is right, and I agree with him, that aging is programmatic, then the fact that we can intervene to increase health span without increasing max life span supports a different conclusion. I hold that there are two distinct programs one that governs the processes of increasing decrepitude and another that can best be described as a death program that kills even healthy individuals within a relatively fixed period of time.
I hold that CR, Metformin, Rapamycin, NR, etc act to slow or eliminate the processes of decrepitude which results in increased health span. The “Decrepitude Program” constitute the first of these two programs.
Regardless of the aforementioned interventions that maximize healthspan, species specific maximum lifespans still exists which are mediated by a death program”. This program kills the individual when the clock built into this second program, the “Death Program” runs out regardless of the deployment of the standard healthspan increasing interventions.
This death program I believe, as Josh has described in previous posts, could be located in the hypothalamus. As Josh has pointed out, work has recently been re-initiated in this area after a period of near dormancy since the work of D. Denkla.
Kevin
You’re right that we know of nothing that can increase human maximum lifespan, but that’s only because it takes a human lifetime to find out. It may well be that metformin or rapamycin increase maximum lifespan. It is probable that CR increases human maximum lifespan. We just can’t know that without many decades of experimentation on thousands of people.
I wouldn’t give up on telomerase just yet Josh. As you know, larger animals express less telomerase than do smaller animals, presumably as a cancer protective mechanism, it’s also very likely true that telomerase expression by itself doesn’t correlate well with a longer lifespan. However, I think that the message of the naked mole rat is that if you have increased telomerase expression combined with an excellent cancer protective mechanism then that combination may well lead to extreme longevity.
A study was done on mice who were engineered to be cancer resistant who had TERT over expression and they lived 25 to 40 % longer.
So maybe before we throw the baby out with the bath water, we should consider combining the cancer protection of rapamycin with something like TA 65 to get the best of both worlds.
Maybe this isn’t an either or proposition.
Just a thought.
Here is that study https://www.ncbi.nlm.nih.gov/pubmed/19013273
It would be interesting to redo that study on mice and track epigenetic changes (assuming we have a equivalent of Horvath clock for mice).
I kind of share Josh’s concern about telomerase accelerating epigenetic changes. However, there must be a way to over-express telomerase while rejuvenating epigenetic since this is what happen during epigenetic reprogramming.
So my hope is that a deeper understanding of epigenetic reprogramming will ultimately lead us to some ways to both rejuvenate epigenetic while still lengthening telomeres.
It would be interesting to redo that study on mice and track epigenetic changes (assuming we have a equivalent of Horvath clock for mice).
I kind of share Josh’s concern about telomerase accelerating epigenetic changes. However, there must be a way to over-express telomerase while rejuvenating epigenetic since this is what happen during epigenetic reprogramming.
So my hope is that a deeper understanding of epigenetic reprogramming will ultimately lead us to some ways to both rejuvenate epigenetic while still lengthening telomeres.
So Mark is going to love this: a recent study published in Nature: So how does our liver regenerate? Via telomerase expressing liver cells!
Hi Josh,
Yes I agree with you in a formal scientific sense that we would need life long human experimentation. But at an intuitive or anecdotal level it seems that max lifespan is not being extended.
People have been practicing CR and the Wisconsin longitudinal study on CR in monkeys has been conducted, neither have, to my knowledge, shown signs that max life span is being extended. Also rapamycin and metformin have been used by people for a long time and I have not heard any indication that there are signs that these people are on a trajectory to extended max life span?
Josh do you think, as I do, that a rule of thumb could be stated as “the longer lived the species the less its maximum lifespan is extended by the interventions such as CR or its mimetics”? It seems that animals that live 30 years or more do not extend max lifespan via the current interventions? This limitation seems most strongly expressed in mammals.
I am not a pessimist for extended max life span in humans, I am just in the camp that a death program is part of the bodies developmental program, if this is correct metabolic interventions will not increase max life span in animals that are long lived and as such have a slow rate of metabolic senescence already.
Generally I agree with you Kevin, we need not only slow down the clock but turn it back – however rapamycin does to seem to do that to some limited extent, and people have not been taking it for a long time (intermittently, as needs to be done for Life extension). Used weekly I think the longest users are at about the 2 year mark. So I would not be surprised to see a max lifespan extension from this, albeit a small one, with a larger effect on health span.
For most of us, increase health span also increase life span as diseases kill us. Increasing human maximum life span seems to be extremely difficult as nobody has thus far broke the record set by Jeanne Calment over 20 years ago.
I agree with Aldebaran that we need a fuller understanding of how telomeres effect epigenetic changes. I suspect we’ll find out that the little bit at the end of the chromosomes (telomere) is vital to the stability of the entire chromosome, and it’s erosion is key to its loss of health.
Btw I suspect the ‘wall’ of max lifespan in humans is likely to be caused by loss of the telomeres in stem cells. Health span is probably controlled partly by your stem cell telomere reserve, and partly by how well you can cope due to your specific genetics as your cells become increasingly decrepid. This is likely where CR, rapamycin, and improved mitophagy from NAD is primarily acting.
Thanks for the tip Akshay, I’ll look that paper up
It is suspected that many super-centenarians died from senile systemic amyloidosis (caused by accumulation of normal transthyretin in the heart and other organs). However, I am not sure why this happen in the first place so maybe it has something to do with stem cells.
To my knowledge, there is no treatment yet but I am wondering if the RNAi approach could help few of them to break the 120 barrier.
Hi Aldebaran, yes I had read that about the transthyretin amyloid build up in centenarians – but let’s not forget these elders are not ‘pictures of health’. Would a otherwise youthful person ever build up such deposits to a pathological level, and if so, how long would it take? My guess is a very long time. You also have the issue that keeping old people living, by say, an amyloid immunotherapy as has been proposed, just sets them up to be killed by the next thing to reach pathological levels. Better to reset gene expression to youthful levels and then deal with anything that comes up rather than try and play wakamole with every metabolic waste product.
Hi Mark,
Totally agree that RNAi (or any efficient amyloidosis therapy) would just alleviate one issue in old people and in the best case allow them to live few more years in better health. But this is something that might be available in few years and that would still be a huge gain for these people.
The long term plan is of certainly to address the root causes of aging as you suggest (for example resetting gene expression to youthful levels) but I doubt that this will be available before at least two decades.
But resetting telomeres to their youthful level does restore gene expression.
Mark,
To my knowledge, gene expressions are mostly controlled by epigenetic changes. I am not sure if restoring telomeres without rejuvenating epigenetic would restore gene expression to youthful levels. Have you read some papers suggesting that?
It has been discovered that as we age CD38 is the main consumer of nuclear NAD+. The reduction in NAD+ causes a shortage of SIRT3. This causes aging via mitochondrial decline. Reference: “CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism” http://www.cell.com/cell-metabolism/fulltext/S1550-4131(16)30224-8 We can reduce nuclear CD38 to youthful levels with apigenin and quercetin, which should restore our sirtuins and increase lifespan Reference: “Flavonoid Apigenin Is an Inhibitor of the NAD+ase” https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3609577/
Apigenin has a half-life of 90 hrs, quercetin of just a few hours. We don’t want to over suppress CD38 – see the health problems of CD38 KO mice. I am taking 250 mg of apigenin once a week. Although quercetin would help reduce CD38, I wouldn’t take much quercetin because of health risks associated with continuous levels.
Hi Aldebaran
Well telomerase certainly isn’t shortening the lifespan of the naked mole rat. Anything but, I would say.
Hi Paul,
Maybe I have missed that point. Is the naked mole rat known to express more telomerase than other rat species? If yes, that might give us some clues about the importance of epigenetic changes.
But so far at least in humans, the high correlation between Horvath clock and mortality suggests to me that epigenetic changes are at least strongly correlated with aging (if not causally implicated). Thus, I would be careful about anything that accelerates the Horvath clock until an experiment has shown some kind of divergence between aging and Horvarth clock (or an equivalent clock for other mammals).
Of course all of that is just speculation at this point. I am betting on the Horvath clock but I might be wrong.
That’s why it would be interesting to reproduce the Blasco study on mices while monitoring their epigenetic changes.
Actually my bad, nmr’s do maintain their telomeres via the alt pathway and not telomerase. Primarily they seem to protect themselves against critically shorts.
I’ve said this before and I’ll continue to say this: the Horvath clock is just measuring how long cells have been around for. That’s its correlation with age. More telomerase means cells last longer. Hardly harmful, but this is being confused with old people having cells that hang around longer because stem cells are exhausted.
Beware of people applying statistics and models to the real world and thinking they understand it.
Hi Mark,
But in that case, shouldn’t we expect rapamycin or CR (which I think slow down stem cells turnover) to accelerate the Horvath clock? However, the opposite is happening.
In both a CR body and a young body cells last longer. But there is an essential difference in that CR slows down metabolism of cells, whereas telomeres restores metabolism to (higher) youthful levels. That explains the difference according to the Horvath Clock.
Re: what you were asking about gene expression, we know from many in vitro experiments telomerase restores gene expression to youthful levels. I think Shay and Wright did a good paper on this.
Interesting.
Food for thought:
I know of critters who amp-up telomerase & relenghten telomeres but only during hibernation & diurnal torpor, so that biological time stretches so profound that many, most pathways undergo biostasis, i.e., period of non-aging. And, behold when they spring back to full active life telomerase throttles back. The outcome, healthspan approaches lifespan, & lifespan is, well, off-chart, e.g., what should be a 4-5-years-only living (5-10 g) mouse-size bat lives to +40 years.
It would seem that exogenous administration of telomerase let alone endogenous triggering at the wrong time is bane that “evolution” worked-out long ago, we are simply to darn slowww [sic] to catch on. I believe Orgel’s second rule may be in order:
“Evolution is cleverer than you are”. – Leslie E. Orgel
which, herein would imply that the process of natural selection though not in itself perhaps intelligent, clever or purposeful, its products are ingenious. Time to tap it & get ingenious h turn the bane into a blessing as the solution is probably lying in plain sight.
No doubt evolution is cleverer than we are. The question is whether evolution was trying to make the animals live as long as possible, or whether she was using telomerase to take control of the lifespan in order to be appropriate for the animal’s ecosystem. This is the “Demographic Theory of Aging”, explained with all the background and reasons most evolutionary biologists have a hard time with it in my book.
Bought & read your book
Learnt things I did not know … thanks 🙂
Since we’ve unique access to torpid-capable “critters” we’re going to see what we can gleam from it in terms of telomeres, telomerase & methylation clock. Said another way, can we extend the bounds of our ecosystem
JIM,
Indian Yogis and some Tibetan monks seem to be able to induce torpor/hibernation and some do live longer. I wonder how much research has been done on these people
Some Aboriginal Australians. Here’s an interesting observation made by officers of The First Fleet to Australia:
“They seemingly enjoy uninterrupted health, & live to a great age. … We have seen them grey with age but not old.”
– Worgan (Fleet surgeon), & Tench (officer), The First Fleet (1788)
A torpor capability is the one universally shared trait common to all “off-chart” long-living animals, mammals. Even things that don’t seem torpid technically are, including, fossorial (e.g., NMR) & diving (e.g., bowhead whale) critters; torpor does not preclude limited/restricted physical activity. Classic telltale hallmarks: spontaneous sub-basal metabolic rate, depressed vitals (including core/periphery temperature); spontaneously reversible; vitals lag follow metabolic rate shifts.
The human side of it: working on it presently, & certain aspects are far down the runway.
😉
Very interesting. Look forward to hearing more.
If you actually want to known, in a nutshell, basically & without going into the details too much, the metabolic slow-down of torpid states masks several unappreciated features.
Firstly, it is known that life-span, t ∝ ~ (body-mass)^0.2, i.e., bigger-sized animals generally live longer. Obviously, you can’t stretch time by up-sizing to whale size. Or can you!? Well, you kinda can. Torpor is special because it transforms the higher pound-for-pound metabolic rate of a small-sized animal to that of larger ones, talking 5 g bat to 200 ton whale, like really. In effect torpor transforms, temporarily small & short-living things into big & long-living things but without the actual body-mass problem. How is this even remotely possible? The thing is, body-mass is not just some inert body-mass, what we’re actually talking about here is activated body-mass, i.e., enzyme-active mass, & enzyme activity in torpor is largely & profoundly depressed. Indeed, mice are more metabolically active, pound-per-pound, than whales, except in torpor, in which case they transform into whales, kinda, sorta, which explains, in part, how such wonders as 29 year old instead of 8 y.o. hibernating/estivating & diurnally torpid 150 g jungle-primates even exist; 29 is the lab version, the wild versions might probably live longer. Of course, small critters arouse more frequently than, say, bears, & they are active for at least a third to half the time, which sandbags their scope for living in slo-mo & as long as a whale; arousals are also extremely costly, metabolically speaking. Mole-rats live continously sub-BMR (Q10 temperature effect ensures it), so they’re continously “torpid”.
But here’s the other thing about torpor, it doesn’t just dilate biological time, it can also paradoxically speed it up. It’s quintessentially-ingenious. If you power-down metabolic rate, temperature generally falls and life’s normal degenerative pathways slow or temporarily arrest, but when temperature falls some enzyme, “cold-loving” enzymes, ones that would be largely inactive at c. 37°C come online & to life. And, this is when the metabolic magic happens, e.g., DNA double-strand break can be repaired, telomere can be prolonged probably without telomerase running havoc on mythelation (?). It seems that this effect (there are others) would result in power-shift (& better balance) between degeneration & regeneration. Think, continuous house-keeping (cleaning & repairs) rather than a mere spring-clean or worse, none. This would explain why animals that live in a slo-mo but still active world live very long & practically healthy lives until they get hit by a bus or infection.*
So, I think, the trick is to run cool & “lights-out” or “slow-mo” torpid or pop in-&-out of it selectively, depending on needs & wants, like Brandt’s (40 y.o.) bat, which should only live at most about 4 years. But how to do that? This is where it gets tricky, & what’s presently being worked on, in humans; we’re cut the chase types & not much into those fat little white liars & tellers of tall & long tails [sic], however awesome.
* Torpor suppresses many, many otherwise extremely lethal pathogenic infections (viruses, bacteria, protozoa, even cancer); not so good with fungus.
So cold immersion with a beta blocker? Maybe some meditation and exercise thrown in ?
Noop. Torpor is an orchestrated event, from within, with various environmental triggers that must be activated in the right sequence. One can’t throw ice-cubes or drug mimics & think it’ll play possum, but many, including DoD (DARPA – Biostasis 03/2018) still think so; not saying cold doesn’t help, has to be properly sequenced & applied. “Ummm” wont cut it either, not really; the EEG flatlines.
Here a yogi was able to lower his metabolic rate: “Voluntary hypo metabolism in an Indian yogi”
https://www.sciencedirect.com/science/article/pii/030645658790060X
Would that be considered at least partially torpor state?
No, that would be considered fully torpid, just not the deepest attainable nor the longest. The definition of torpor merely requires a metabolic rate that falls below the normal basal rate; the degree of corezcooling depends on the metabolic rate & several other factors. You’d be hard-pressed to detect EEG in deepest torpor as it’s ultra-low voltage. The thing to note is that torpor is not some particulate metabolic rate or core temp or physical activity level but spans a spectrum, which becomes more resolute & easier to detect in terms of vitals the lower these signatures. Consciousness can be maintained to ~ 29 – 30°C but with considerable cognitive & physical deficeit. Even upon reaching merely 33-34°ish, e.g., slurred speech, muscle stiffness, much suppressed vitals, time-warpping, etc. (pers. obs)*. In that sense, it’s like hypothermia, but the anapyrexia (i.e., down-regulated cooling) of torpor is, however, not pathological like (accidental or “therapeutic”) hypothermia, it’s spontaneously reversible as you can see from the link; hypothermia isn’t reversible without external rewarming assistance, though the reversibility & pathogenesis of hypothermia is, admittedly, time-dose dependent & hence why it can to a certain extent protect/preserve life for some time. BUT, animals don’t medidate (as far as I know) & neither do the torpid human experiments I’ve been involved with, but that is not to say it precludes entry via meditation.
* http://img1.wsimg.com/blobby/go/13edc45c-64c6-44b8-9675-85299f8f01da/downloads/1c9nmms46_667093.jpg
Thank you JIM for the explanation. I checked up your website. I guess to find out how your experiment works, I have to go through the gateway. Though I think I know the answer to the first puzzle (bear-cave or bat cave), I don’t feel smart enough nor brave enough to go search for that relevant someone up the river, deep in the jungle.
Sorry, I didn’t answer your question. We’d be talking Big & deep systems bio, meaning, sharp-edged double-edged, but we know we can sit on this forever & it wont stay buried, just trying to roll it out in a controlled & responsible way, i.e., slow-like. I think Ralph Ellison captured it best:
“Please, a definition: A hibernation is a covert preparation for a more overt action.” – The Invisible Man (1952).
PS: I’m not into covert nor overt action, or invisible men … they tend to be psychopaths generally anyway.
There’s little risk, in that the system will reboot & rewarm, but if one is thoughtless enough to do it where one shouldn’t then it’s like standing in the middle of the road long enough; animals look for a safe place & some non-human primates bury themselves alive, in the jungle floor, for 7-8 months straight.
Well, I wish you success in your endeavours.
It seems that stem cells and progenitor cells thrive with excess telomerase, whilst lack of telomerase causes cancer cells to go into senolysis.
This seems like a catch 22. However, it is a similar situation to the feeding-fasting cycle. Our cells need food, yet periodic fasting brings about a cleansing and senolysis.
Therefore, a possible strategy would be to take a telomerase inhibitor first, to drive cancer cells to senolysis, then take a telomerase activator to invigorate stem and progenitor cells.
And repeat this cycle.
This strategy rests on the idea that cancer cells are more vulnerable to senolysis than healthy cells when exposed to a telomerase inhibitor . If they are, then this cycle should magnify the good cells.
Telomerase inhibitors include curcumin, milk thistle…
Telomerase activators include cycloastragenol, ginkgo bilbao, resveratrol, genistein, DMSO
So, I would propose a cyclic protocol, similar to the feeding-fasting cycle
Ref: https://www.intechopen.com/books/enzyme-inhibitors-and-activators/telomerase-inhibitors-and-activators-pharmaceutical-importance