
Can methylation clocks be relied on in this context? The earliest clocks were trained on chronological age only, and yet they predicted morbidity and mortality better than chronological age. I’ve been enthusiastic about the technology since 2013. But recently there have been substantial challenges to the validity of all the existing clocks.
An article published last week fulfills a wish I’ve expressed in previous columns. I’ve believed that methylation clocks are the best tool that we have for evaluating anti-aging interventions, and there is potential to accelerate anti-aging trials enormously. But it’s not proven that setting back any given methylation clock will result in added longevity.
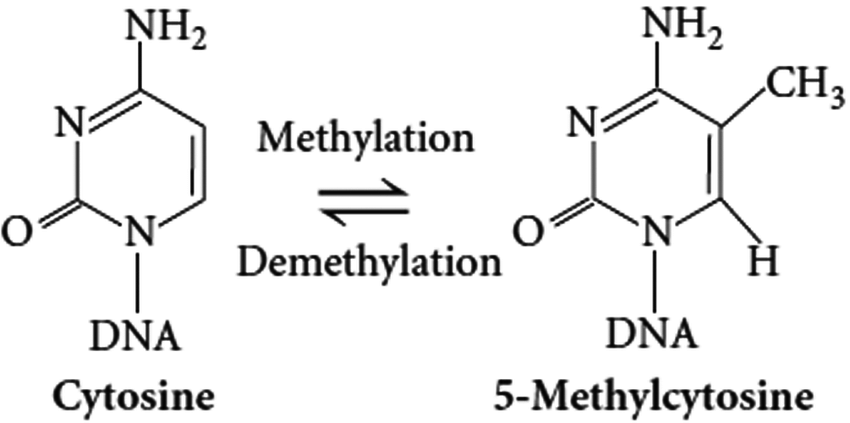
Methylation clocks are a surrogate for gene expression. The problem is that two things happen to gene expression with age.
[1] Programmed aging turns on chronic inflammation and shuts down repair mechanisms with age.
[2] The body is increasingly damaged, and repair mechanisms are triggered in response to that damage.
Yes, the body is fighting with itself as we get older. Some of the epigenetic changes that happen are suicidal, but the fundamental self-protective responses are still in play.
Suppose you take an anti-aging pill. If the pill makes you score younger according to [1] then you’re destined to live longer; but if you score younger according to [2] the chances are your life will be further shortened.
The way most methylation clocks are derived gives you no indication what mix of [1] and [2] goes into the age algorithm. To complicate the situation further, a strong type [2] response might be an indication of more damage, which would be bad, or an effective hormetic response, which would be good.
(I have argued that the PhenoAge clock is probably biased toward [1], and I have worried that the GrimAge clock may have a lot of [2].)
This preprint by Ying et al, a collaboration between the Gladyshev lab at Harvard and the Horvath lab at UCLA, uses bioinformatic methods to confront the problem head-on.
“It is unclear whether the DNA methylation changes that are used to predict age are causal to aging-related phenotypes or are simply byproducts of the aging process that does not influence aging themselves.”
I would add: Or worse — some methylation changes with age actually mitigate damage [2], and “resetting” those would be damaging to health, probably shortening lifespan.
The experiment we want to do would be to change methylation at thousands of CpGs in a sample of people and see how the change affects their health. This is way impractical, both because we cannot yet manipulate cytosine methylation directly, and because the number of subjects involved would be enormous. A practical approach would be to calibrate an aging clock against historic data using remaining lifespan as a target (see below, “A Simpler Alternative”). This procedure has not been followed, to my knowledge, but PhenoAge, GrimAge, and Dunedin Pace all have some features of this procedure.
The new article that I’m reviewing here takes a different approach, deploying GWAS.
GWAS (Genome-Wide Association Studies) have been used in the past to correlate various health outcomes with common genetic variants. For example, one could look at 23-and-me data for people who have had heart attacks and compare to a matched sample of people who have not had heart attacks; then look for genome variations (SNPs) that tend to be different in the two groups. Fast computers can do millions of correlation calculations and report back the ones that show the strongest results.
How might this be applied to methylation sites? Methylation affects which genes are turned on in an individual, rather than which allele of the gene that individual happens to have. The trick is to look for 3-way correlations. MeQTL stands for “methylation quantitative trait loci”. There are SNPs that are associated both with methylation changes at particular CpGs and also with health outcomes. Maybe it is through the CpG that the methylation change is causing the health effect. How can they determine if this is the case? A proposal was put forward by Richardson et al [2018]
Abstract
We have undertaken a systematic Mendelian randomization (MR) study using methylation quantitative trait loci (meQTL) as genetic instruments to assess the relationship between genetic variation, DNA methylation and 139 complex traits. Using two-sample MR, we identified 1148 associations across 61 traits where genetic variants were associated with both proximal DNA methylation (i.e. cis-meQTL) and complex trait variation (P < 1.39 1008). Joint likelihood mapping provided evidence that the genetic variant which influenced DNA methylation levels for 348 of these associations across 47 traits was also responsible for variation in complex traits. These associations showed a high rate of replication in the BIOS QTL and UK Biobank datasets for 14 selected traits, as 101 of the attempted 128 associations survived multiple testing corrections (P < 3.91 1004). Integrating expression quantitative trait loci (eQTL) data suggested that genetic variants responsible for 306 of the 348 refined meQTL associations also influence gene expression, which indicates a coordinated system of effects that are consistent with causality. … Though we are unable to distinguish mediation from horizontal pleiotropy in these analyses, our findings should prove valuable in prioritizing candidate loci where DNA methylation may influence traits and help develop mechanistic insight into the aetiology of complex disease. [Richardson 2018]
The method is to look for 3-way correlations between (A) the trait in question, (B) methylation of a particular CpG site and (C) a point genetic variation (SNP). It’s a dangerous game for a number of reasons. First, scanning over a huge number of possibilities assures that some of them come up positive just by chance. If you run an experiment 20 times, you’ll probably come up with some result (in one run) that shows up as statistically significant, with odds against chance of p<0.05. Similarly, if you search a million CpG sites to see if any of them are associated with your favorite trait, then one of them is bound to show up with an association so strong that you report p<10^-6. It sounds impressive to say p<0.000001, but in this case it’s just the laws of chance operating on a huge number of possible associations.
The people who do these analyses are not incompetent. They know this, and they compensate by setting the threshold 100 times low, in this case p<10^-8. But these probabilities are always computed based on a number of unverifiable assumptions, and it’s always possible that some chance association slips in.
There’s a second problem with these 3-way associations. Remember that what we’re trying to establish is that a particular CpG causes a particular trait.
CpG ⇒ trait
Lacking a way to directly correlate the CpG with the trait, this method relies on the fact that both the trait and the CpG are associated with the same SNP. The “causal” implication is that
SNP ⇒ CpG ⇒ trait
But the authors gloss over the possibility that
SNP ⇒ trait and
SNP ⇒ CpG
In words: Suppose the SNP has two actions (pleiotropy is the rule rather than the exception) so that it independently causes the trait and the CpG methylation. In this case, there is no indication that the CpG actually causes the trait. An even worse possibility is this:
SNP ⇒ trait ⇒ CpG
This is particularly worrisome. For example if the trait is inflammation, you would like to establish the fact that this particular methylation site causes inflammation. But if the CpG is associated with a quenching response to inflammation, then you’d expect that CpG activation might be part of the body’s solution to perceived inflammation.
The fact that you tend to see fire trucks and fires in the same places around a city doesn’t mean that the fire trucks cause fires.
I am all for using fancy statistics to learn about the metabolism. But the headier the statistics you use, the more you have to worry about things that could go wrong.
This is all in the context of my limited sophistication with advanced statistical methods. The authors of this study recognize all the hazards that I have catalogued, and they present arguments why they have effectively compensated for them. I don’t have the background to pass judgment on those arguments.
“Here, we leveraged large-scale genetic data and performed epigenome-wide Mendelian Random ization (EWMR) on 420,509 CpG sites to identify CpG sites that are causal to twelve aging-related traits. We found that none of the existing clocks are enriched for causal CpG sites.”
I take this as a red flag. How can it be that there is negligible overlap between the CpGs found by this new GWAS methodology and any of the Horvath clocks of the past? I believe that aging is driven at a deep level by epigenetic changes, and this gives credibility to the idea behind methylation clocks. I have acknowledged the risk that these clocks may be polluted with some CpGs that are not drivers of aging [1] but responses to aging [2]. But if these new results are to believed, we have to throw out GrimAge and PhenoAge as worse than useless.
“Contradicting the popular notion that most age-related changes are bad for the organism, our findings revealed that, in terms of the number of CpGs, there was no enrichment for either protective or damaging methylation changes during aging.”
The first two papers to propose that methylation is a primary driver of aging appeared in 2012 [one, two]. I was an enthusiastic supporter in 2013. A decade later, these papers have seeded an entire field of study. But the above captioned statement suggests that this was all in vain, that methylation changes with age are a correlation only, and they don’t really have to do with either drivers of aging [1] or protection from damage [2].
“Our results suggest that the known lifespan-related effect [of APOE] may be mediated by DNA methylation.”
This in itself is a stunning conclusion, and should lead to methylation-based therapies for the millions of people who are at elevated risk of AD and CVD based on APOE4 in their genomes.
Three new clocks
Ying et al announce the creation of three new methylation clocks based on the science described above. CausAge was developed using traditional methods with an additional boost for sites that are identified through GWAS as associated with causes of age-related decline. DamAge was designed to include only sites that are associated with increasing damage to the body; while AdaptAge was designed to be based on the opposite — sites associated with protective adaptations. Scoring older on the DamAge clock is presumably a bad thing because it indicates more damage, while scoring older on the AdaptAge clock should be associated, paradoxically, with a longer life expectancy. “Therefore, we hypothesized that DamAge acceleration may be harmful and shorten life expectancy, whereas AdaptAge acceleration would be protective or neutral, which may indicate healthy longevity.” I had to read this sentence several times because it was the reverse of what I expected from the text. Based on this sentence, I’m guessing that DamAge corresponds to what I called [1] above, and AdaptAge is [2].
“We found that short-term treatment with cigarette smoke condensate in bronchial epithelial cells significantly accelerated DamAge but did not affect other tested clocks (Fig. 6c). Additionally, a 6-week omega-3 fatty acid supplementation in overweight subjects, which has been shown to be protective against age-related cardiovascular diseases, significantly increased AdaptAge and reduced DamAge (Fig. 6c).”
This is another question mark for me. Cigaratte smoke makes DamAge look older. I have always imagined that the direct effect of smoke is to damage lung tissue, and that some kind of epigenetic response to this is the body’s attempt to protect itself. That would be a type [2] (hormetic) response. But according to the above captioned sentence, cigarette smoke has a direct and detrimental effect on methylation, a type [1] response. I can’t say this is impossible, just unexpected.
A Simpler Alternative
The ideal methylation clock would be rooted in a deep understanding of biochemistry. We would know which genes are associated with inflammation, with immune senescence, with sex hormones, with apoptosis, with blood lipids—and also which CpGs turn these genes on and off. This kind of detail is not available at present, but it is a reasonable long-range goal.
Here’s my proposal for creating an aging clock that measures what we are most interested in: is a given person’s biological age older or younger than others of the same chronological age?
Start with a historic sample of methylation profiles from biobanked samples of people who have died in the intervening years. Train a new methylation clock on the number of years that each subject survived since the sample was drawn, and include in the target a penalty for chronological age. In other words, the training target is the number of years the person actually lived minus the number of years he would have been expected to live given his chronological age.
(After juggling in my mind the gender of pronouns in the previous sentence, I realize that, given what we know about different aging mechanisms by sex, we really should do this analysis so as to create separate methylation clocks for men and women.)
There is a lot more data available now than when Steve Horvath did his pioneering work in 2012-13. His clocks have been constrained to be linear in each methylation site for adults. But we have reason to believe that epigenetic changes come in waves, with different methylation sites being most important at different ages. So let’s calibrate two different clocks for each decade of life, and for men and women.
While I’m making a wish list, I would add that my ideal methylation clock should avoid being too sensitive to any one CpG. This is for the practical reason that there are quality control issues that come from lab technique and from manufacture of bead chips.
The problem comes from having large positive and negative contributions in the age algorithm, which cancel sensitively in the final result. The way to avoid this problem is to derive two separate clocks, one based only on CpGs that increase methylation with age and the other based only on those that decrease methylation with age; then average the two calculations for the plus and minus clocks.
I believe this project is more than feasible, it should be a lot less work than the present analysis by Ying et al. And for me, it would have the advantage of being transparent, not dependent on unnecessary assumptions, and avoiding indirect references from 3-way correlations. If anyone reading this has access to the data, I would love to do the statistics.
And why should we have faith that the construction I outline should lead predominantly to sites that have a causal relationship to senescence [1]? It’s not a slam dunk, but the fact that a person dies later than expected is a good indication that protective responses are up and programmed aging is down. Our understanding of the situation is confounded by hormesis.
The Bottom Line
This new work should cause re-thinking of all the anti-aging work of the last several years that has relied on methylation clocks to gauge success. The message is that the clocks that have been developed so far are at best orthogonal to the real causes of aging, and at worst they could be encouraging interventions that actually shut off the body’s natural protections. If we believe the new results, DamAge would be a far better choice than anything previously available for any anti-aging trial, but the difference DamAge minus AdaptAge might be even better.![]()
As the DataBETA project is launched this fall, I have a direct professional interest in choosing the right technology to evaluate a wide range of interventions and combinations that are commonly used by people who are trying to live longer.
I have been enthusiastic about methylation clocks from the beginning on the basis that (A) for theoretical reasons, I find it credible that epigenetics drives aging, and (B) the original Horvath clock, derived only from chronological age, predicts mortality better than chronological age. I have a hard time throwing that reasoning out based on one new preprint. But there has also been Katcher’s lifespan experiment, and Levine’s caution based on theoretical grounds, both of which have shaken my faith in methylation clocks earlier this year.
Frankly, if Horvath’s name hadn’t been on this ms, I would not have been inclined to take it seriously. Should we believe the new results, which seem to discredit all the analysis that he and others have done in the first decade of epigenetic clocks?
I don’t have the expertise to answer this question. I can hope that this work receives a deep and respectful challenge during the peer review process, and that people with more statistical chops than I have will debate both sides of this question over the coming months.
Discover more from Josh Mitteldorf
Subscribe to get the latest posts sent to your email.
Possibly relevant is a vey recent study about how the people who survived the Black Death have several gene variants that cause a more aggressive immune response, at the cost of a higher frequency of autoimmune diseases:
https://www.nature.com/articles/d41586-022-03298-z
So that’s a clear tradeoff: more aggressive immune system = immune systems misfires and erroneously attacks the self more often.
Those who argue that aging is “the best the organism can do” (as opposed to a programmed, self-destructive behavior) would probably say that if we reverse the methylation clock – and thereby turn repair mechanisms and the full capability of the immune system back on – we will see immediate health improvements combined with a (statistically) lower average remaining lifespan, because the rejuvenated repair mechanisms would cause some sort of ultimately fatal issue (auto-immune issues would be a strong contender).
With this in mind, there may be a dataset that is useful but hasn’t been looked at: skin care procedures like laser treatments or microneedling create micro-damage and this stimulates repair.
Clearly, these treatments can only work because the body had the capacity to completely repair the tissue in question, and chose not to until stimulated by micro-damage.
So do patients that receive these treatments go on to develop a higher incidence of skin-related auto-immune diseases? Rosacea? Or even melanoma?
What about people who have “active release” or “rolfing”? It’s the same concept (micro-damage to stimulate repair) – so do we then see a greater incidence of myofascial auto-immune issues or cancers? Arthritis?
Any luck getting Google’s Captcha tamed?
Great article! Didn’t think of the points you raised when initially reading the study.
I’ve started to think that Dr. Horvath is listed as a coauthor whenever he’s asked to verify CpG results from his website. I wouldn’t ascribe his endorsement to silliness during the last three months such as “Epigenome-wide analysis of DNA methylation and optimism in women and men” and “Epigenome-wide meta-analysis of BMI in nine cohorts: examining the utility of epigenetic BMI in predicting metabolic health.”
Regarding DataBETA:
Are you still looking for subjects?
Excellent analysis by Josh. That’s why so many visit this blog. The CpGs selected by Horvath’s algorithms do not need to be causal of aging nor protective to predict biological age. The changes too at these sites do not need to reflect aging related damage. His algorithm identified particular sites where changes that occur with age are so consistent that a bioage clock can be developed from it. Changes are constantly happening globally since the time the egg is fertilized and continue till we die. It’s just that algorithms picked up locations which collectively change in a pattern that matches chronological age. If an intervention is able to change the location in that pattern that matches a chronological unit that is seen in a younger subject compared to its calendar age that continues to be an incredible change/result regardless of whether those sites are causal of aging or not. If one studies Horvath’s clocks one can see how accurate they remain and how little noise is actually present. The algorithm selects those sites after ‘studying’ millions of sites in thousands of samples. This becomes a clock only when the slider moves consistently over and over again. One can’t argue with maths. Josh you mentioned about our female lifespan study which is still not over. But what about the 30 biomarkers that also matched the Horvath clock? They were biochemical, histopathological and haematological. What about the functional tests like grip strength that showed in a recent study 2.7 times stronger than old control? Lifespan and biological age reversal should not be confused. We know very little to conclude that becoming younger would lead to longer lifespans. Logically it should but Nature may have surprises in store. Anyway that should not become a consideration to lose faith in these wonderful algorithms. We do see some trees live in youth for thousands of years. We can not dismiss this by saying that trees are completely different- we share so many similarities in our core biology- they too have genes, they too have cells, they too have exosomes, etc. so if these trees can live for thousands of years in youth then so should we be able to achieve that at some point.
“If an intervention is able to change the location in that pattern that matches a chronological unit that is seen in a younger subject compared to its calendar age that continues to be an incredible change/result regardless of whether those sites are causal of aging or not. ”
Fully agree with reasoning.
Hello
RE: Paper #1 from 2012 with lead author Joao Pedro De Magheles that was the “first” to describe how DNA methylation is involved in aging…
At an aging conference in Spa Belgium in the year 2000 I handed a copy of my 1998 paper to Joao that went on and on about how DNA methylation / demethylation was what controlled aging.. that was about 35%+ of the 50 page paper titled the Evolution of Aging – A New Approach to an Old Problem of Biology..Med Hyp Sep 98 He was just a grad student at the time…I told him ..you can take this and run with it as I have to get back to work in the business world (real estate)… Looks like he did run with it! 14 years later HAHA
It is very interesting that it seems long lived bats appear to make use of incomplete epigenetic rejuvenation, and this might be causal to their great longevity over rodents.
>“We knew that, but we didn’t know if we would detect changes in epigenetic age due to hibernation.” The researchers determined that hibernating over one winter extends a big brown bat’s epigenetic clock—a biological marker of aging—by three-quarters of a year.-https://today.umd.edu/briefs/hibernation-slows-biological-aging-in-bats-umd-led-study-finds
I do believe these changes are causal, and extend their lifespan. And as you’ve suggested aging is epigenetically controlled.
Sounds like the bats have something equivalent to Yamanaka factor usage without the risk of de-differentiation.
Josh, your interpretation of various age clocks over the years has very much helped me understand them better, thank you.
This has been particularly useful to inform the epigenetic aging part of an online video longevity course I’m working on.
My personal experience (n=1) with such clocks are inconsistent. I “score” 18 years younger biologically with the PhenoAge test, but 1 year older with the TruDiagnostic epigenetic test offered by AgelessRX that is based on the DunedinPACE algorithm.
The study you speak about in this post was eye-opening, as I had not considered how methylation of certain genes could be determined to be pro-aging when in fact they’re protective. If I recall correctly, the study cited IIS and GH downregulation as examples of protective measures that occur as we age. These two anabolic hormones should be upregulated when young/youngish, but are pro-aging when we get older when we need more catabolic activity to focus not on growth but repair.
jg
The inconsistent results you mention, remind me of similar comments from others on this blog. All of these companies have sprung up, each offering their own proprietary methylation analysis, sometimes coupled with other blood test data and lifestyle questionnaires. Isn´t there anybody who offers a ¨plain vanilla¨Horvath test ?
Gregory, a company named Novos has a bioage test based on AI interpretation of your face. Just upload a recent photo. They claim some degree of accuracy. I wrote about it and other bioage tests here: https://garmaonhealth.com/discover-your-biological-age-with-these-4-tests-part-1/
Irrespective of what a test says, I take solace in how I look, feel and can physically perform. A youthful look (relative to chronological age), feeling energetic and emotionally stable plus maintaining cardiorespiratory fitness, strength and lean muscle indicate that a person is aging well.
This is the website for calculating age from your face
https://novoslabs.com/faceage/#test
Novos says that my face is 60 years old and my eyes 48, but my wrinkles put me in the 90s.
Out of curiosity, I did the test too:
Facial age: 52
Eye age: 41
Eye bags: 66
Facial Wrinkles: 56
Facial uniformity: 37
My chronological age is 67.
Oh, forgot to mention that this new study Microscopic Imaging of Epigenetic Age is different than any other that I’ve read:
“We have developed a novel approach, microscopic imaging of biological age (miBioAge), which computes multiparametric signatures based on the patterns of epigenetic landscape in single nuclei. We demonstrated that such miBioAge readouts can robustly distinguish young and old cells from multiple tissues, reveal aging progression in peripheral blood (e.g. CD3+ T cells), and reflect changes of epigenetic signatures consistent with expected slowdown or acceleration of biological age in chronologically identical mice treated with caloric restriction or chemotherapy. Because miBioAge readouts are computed from individual samples without applying linear regression, we posit that this novel biomarker may provide personalized assessment of functional aging.”
https://www.biorxiv.org/content/10.1101/2022.10.16.512441v1.full.pdf+html
“Turbuckle” uses TruAge on the Longecity forums.
As everyone ages, and there are no specific SNPs for aging, using SNPs to correlate with methylation in the hope that this will inform an understanding of aging is a fruitless errand.
Also, as there is so much more DNA code than recognisable genes, most methylation has no obvious relationship with known SNPs.
Methylation clocks are useful, we know that by looking at how methylation changes from embryonic to stem cells to disposable somatic cells.
But we need to take a step back and look at what is actually physically happening to cells. Not bury our heads in statistics.
Blank comment so that I get updates of other comments.
I guess these clocks can give you some indication of how you are aging compared to others but I think most people are still waiting for that magical pill that you can take and months later you truly look 10 – 15 years younger. I have seen some pretty big results with some clocks but the outer appearance usually never measures up to the clock reversal in years though I have seen a few cases I would say the person really did look about 5 years younger or so. Not sure if humans just don’t get the same bang for the buck with these aging treatments like mice and rats do or what is up with that. Part of it may be many trials using dark colored mice where the improvement with less greying and thick coats really stands out compared to the non-treated mouse that has lost a lot of hair and is greying. My hunch is Katcher’s E5 is probably the strongest stuff tested as of now but since he used those white rats you didn’t get the super crazy contrasts with the treated rats like you would with the dark mice. Maybe when they start producing a good amount of E5 Katcher will test it on his facial skin to see what it does like he did with his hand.
It would be particularly interesting to see what happens if he applies it to his scalp since most men start losing hair in their 20s.
Which reminds me, a little off topic, but if the proteins in E5 can be extracted from animals and isolated instead of manipulated then the inhaled version of E5 (something was said about that possibility being explored), I wonder if it might be able to pass FDA approval entirely as some sort of food product or nutritional supplement. Maybe it’s better or only possible to get it through clinical trials, it may be an important option for them to have at their fingertips.
I personally like to use the GeroSense app to compare with other measurements. If you are not moving around like a younger person after an intervention you likely haven’t helped your age and Gero will detect that.
Josh,
I think you will find this video interview of Peter Fedichev informative. He has a background similar to yours so he thinks about aging from a different viewpoint.
Dr. Peter Fedichev, PhD – CEO, Gero – Hacking Complex Diseases & Aging with AI & Digital Biomarkers
A very recent study – Nov. 2 – announces ‘A generalizable epigenetic clock captures aging in two nonhuman primates’.
An abstract follows: “Epigenetic clocks generated from DNA methylation array data provide important insights into biological aging, disease susceptibility, and mortality risk. However, these clocks cannot be applied to high-throughput, sequence-based datasets more commonly used to study nonhuman animals. Here, we built a generalizable epigenetic clock using genome-wide DNA methylation data from 493 free-ranging rhesus macaques. Using a sliding-window approach that maximizes generalizability across datasets and species, this model predicted age with high accuracy (± 1.42 years) in held-out test samples, as well as in two independent test sets: rhesus macaques from a captive population (n=43) and wild baboons in Kenya (n=271). Our model can also be used to generate insight into the factors hypothesized to alter epigenetic aging, including social status and exposure to traumatic events. Our results thus provide a flexible tool for predicting age in other populations and species and illustrate how connecting behavioral data with the epigenetic clock can uncover social influences on biological age.”.
Link at biorxiv: https://www.biorxiv.org/content/10.1101/2022.11.01.514719v1
A very interesting paper for accurately predicting remaining lifespan for mice, utilizing machine learning techniques and approaches borrowed from the dynamic systems theory.
https://www.nature.com/articles/s41467-022-34051-9